
(S,S)-2-(((Hydroxynaphth-1-yl)(4′-nitrophenyl)methyl)amino)-3-methylbutanoic Acid Methyl Ester


Abstract
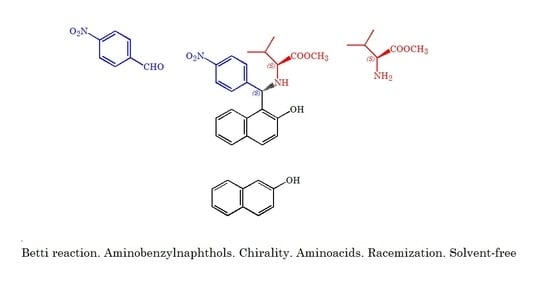
{kind=link}
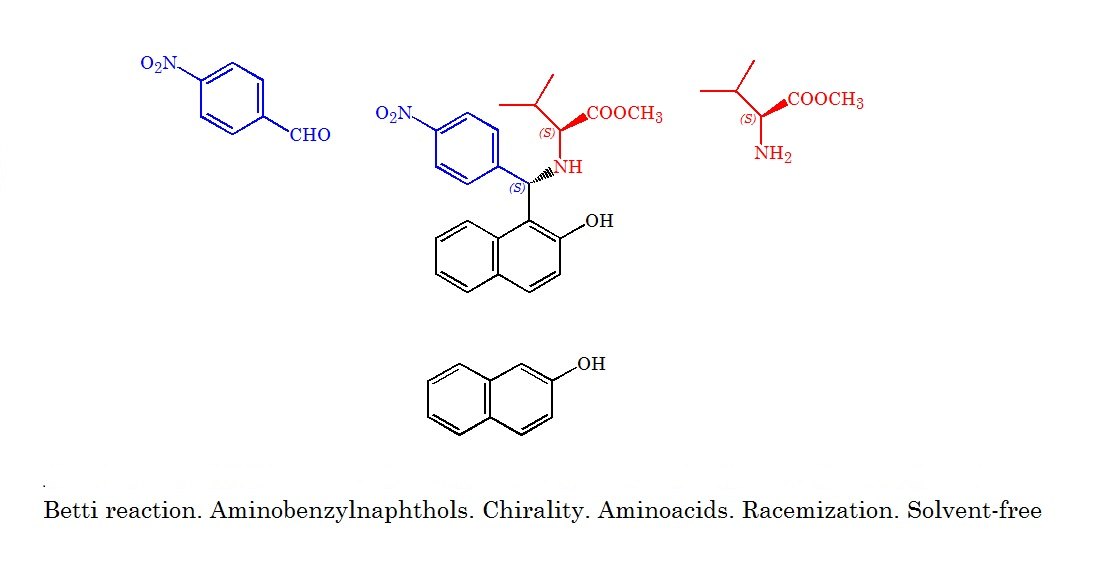
{kind=link}
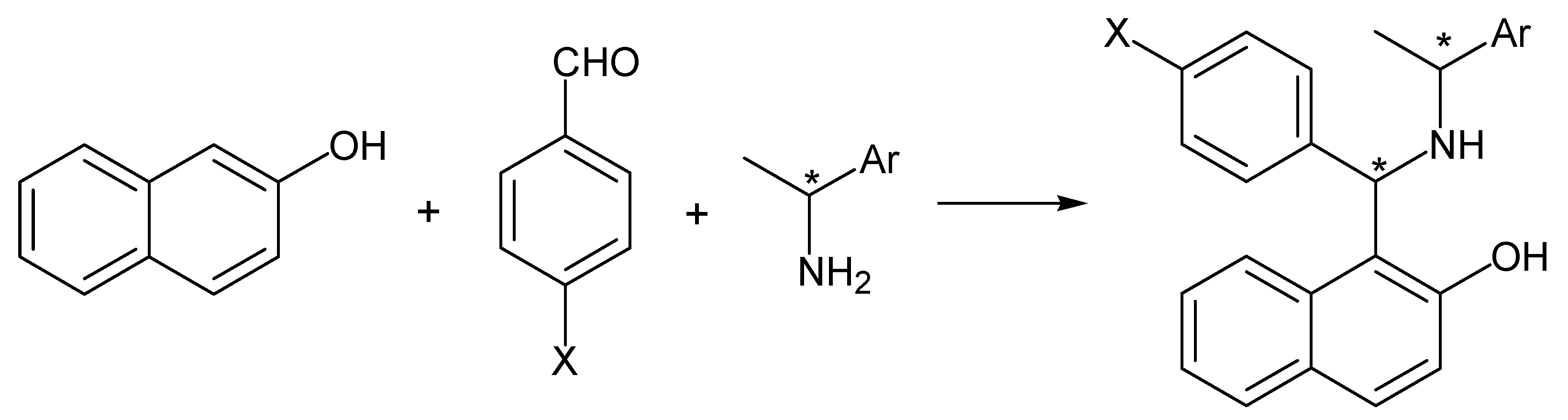
{kind=link}
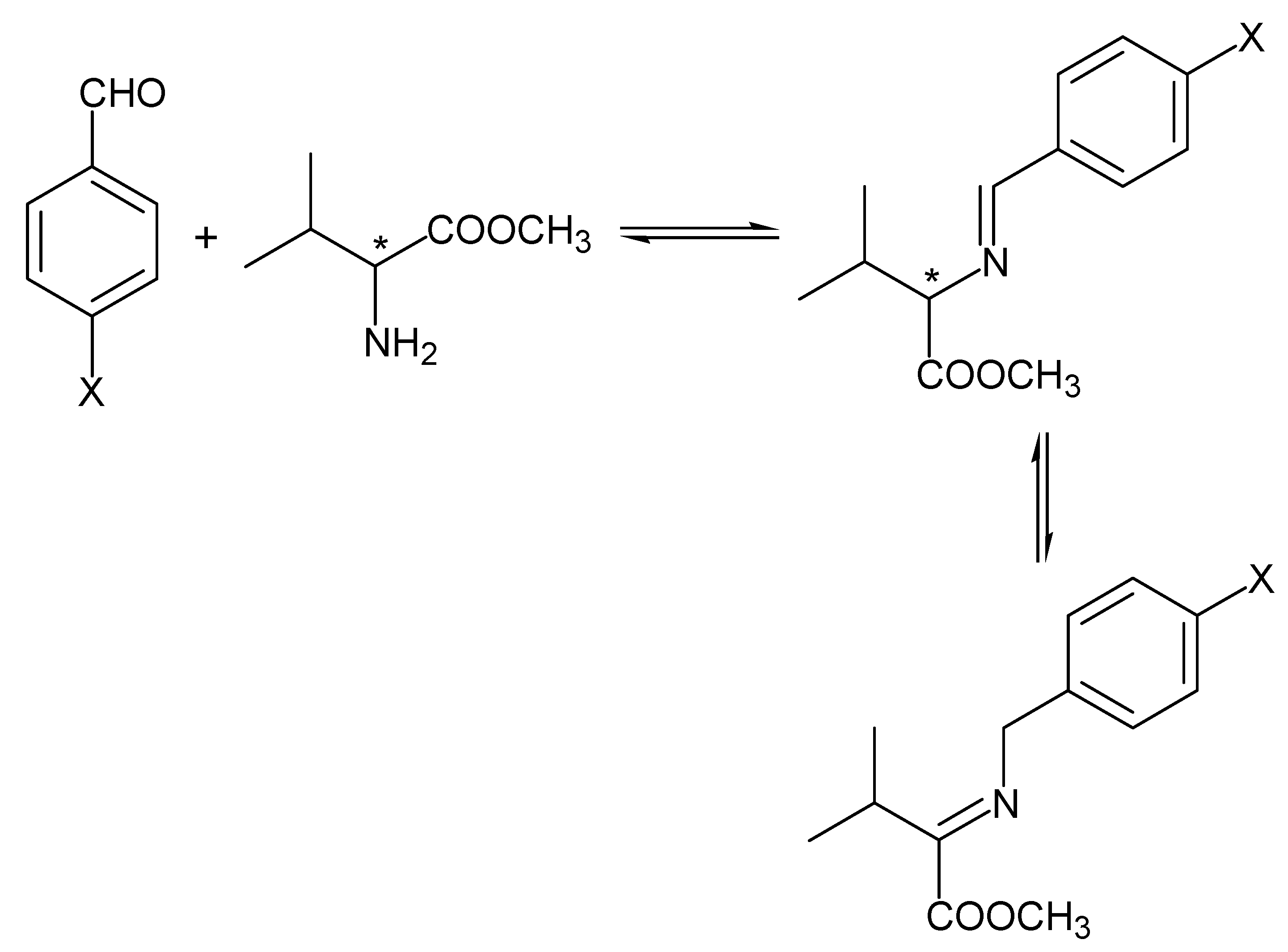
{kind=link}
1. Introduction
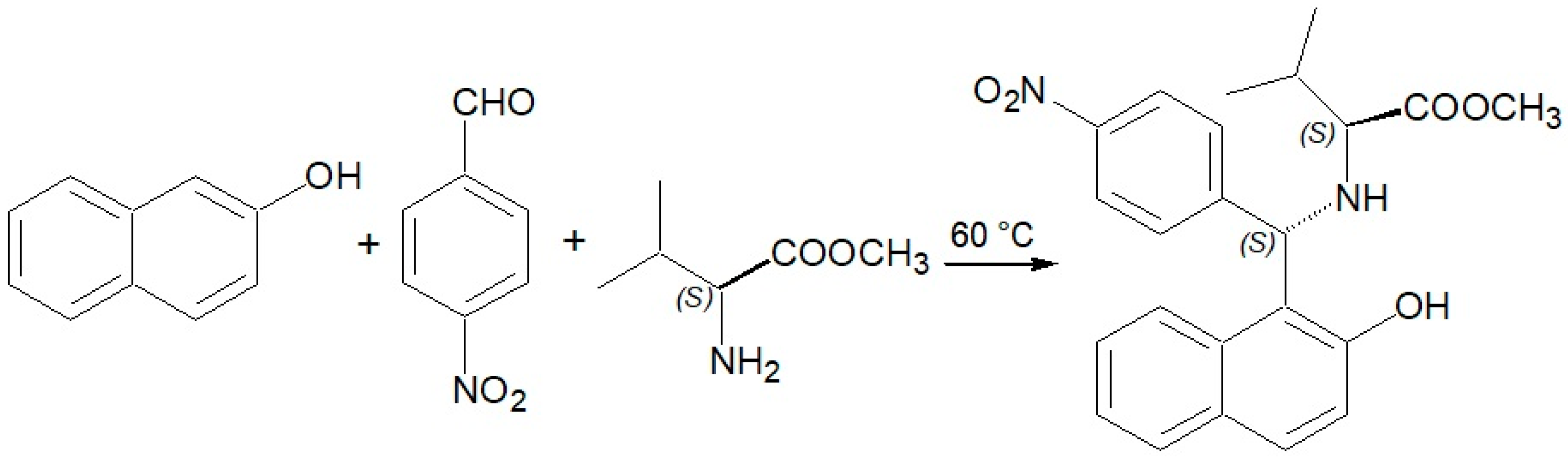
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Cardellicchio, C.; Ciccarella, G.F.; Schingaro, E.; Scordari, F. The Betti base: Absolute configuration and routes to a family of related chiral nonracemic bases. Tetrahedron Asymmetry 1998, 9, 3667–3675. [Google Scholar] [CrossRef]
- Cardellicchio, C.; Ciccarella, G.; Naso, F.; Perna, F.; Tortorella, P. Use of readily available chiral compounds related to the Betti base in the enantioselective addition of diethylzinc to aryl aldehydes. Tetrahedron 1999, 55, 14685–14692. [Google Scholar] [CrossRef]
- Naso, F. Mario Betti: A Giant in the Chemistry Scenario of the Twentieth Century. Substantia 2017, 1, 111–121. [Google Scholar]
- Cardellicchio, C.; Capozzi, M.A.M.; Naso, F. The Betti base: The awakening of a sleeping beauty. Tetrahedron Asymmetry 2010, 21, 507–517. [Google Scholar] [CrossRef]
- Iftikhar, R.; Kamran, M.; Iftikhar, A.; Parveen, S.; Naeem, N.; Jamil, N. Recent Advances in the green synthesis of Betti bases and their application: A review. Mol. Divers. 2022. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, M.A.M.; Cardellicchio, C.; Magaletti, A.; Bevilacqua, A.; Perricone, M.; Corbo, M.R. Bioactivity of a Family of Chiral Nonracemic Aminobenzylnaphthols towards Candida albicans. Molecules 2014, 19, 5219–5230. [Google Scholar] [CrossRef] [PubMed]
- Mallamaci, R.; Capozzi, M.A.M.; Cardellicchio, C. Antiproliferative Activity of Aminobenzylnaphthols Deriving from the Betti Reaction. Appl. Sci. 2022, 12, 7779. [Google Scholar] [CrossRef]
- Capozzi, M.A.M.; Cardellicchio, C. Stereoselection in the Betti reaction of valine methyl esters. Tetrahedron Asymmetry 2017, 28, 1792–1796. [Google Scholar] [CrossRef]
- Capozzi, M.A.M.; Alvarez-Larena, A.; Piniella Febrer, J.F.; Cardellicchio, C. A combined structural and computational investigation of aminobenzylnaphthol compounds derived from the Betti reaction using valine methyl ester. New J. Chem. 2021, 45, 20735–20742. [Google Scholar] [CrossRef]
- Cardellicchio, C.; Capozzi, M.A.M.; Alvarez-Larena, A.; Piniella, J.F.; Capitelli, F. Investigation on the weak interactions assembling the crystal structures of Betti bases. CrystEngComm 2012, 14, 3972–3981. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capozzi, M.A.M.; Cardellicchio, C. (S,S)-2-(((Hydroxynaphth-1-yl)(4′-nitrophenyl)methyl)amino)-3-methylbutanoic Acid Methyl Ester. Molbank 2022, 2022, M1528. https://doi.org/10.3390/M1528
Capozzi MAM, Cardellicchio C. (S,S)-2-(((Hydroxynaphth-1-yl)(4′-nitrophenyl)methyl)amino)-3-methylbutanoic Acid Methyl Ester. Molbank. 2022; 2022(4):M1528. https://doi.org/10.3390/M1528
Chicago/Turabian StyleCapozzi, Maria Annunziata M., and Cosimo Cardellicchio. 2022. "(S,S)-2-(((Hydroxynaphth-1-yl)(4′-nitrophenyl)methyl)amino)-3-methylbutanoic Acid Methyl Ester" Molbank 2022, no. 4: M1528. https://doi.org/10.3390/M1528
APA StyleCapozzi, M. A. M., & Cardellicchio, C. (2022). (S,S)-2-(((Hydroxynaphth-1-yl)(4′-nitrophenyl)methyl)amino)-3-methylbutanoic Acid Methyl Ester. Molbank, 2022(4), M1528. https://doi.org/10.3390/M1528