N4-(2-Amino-4-fluorophenyl)-N1-(3-{2-[2-(3-{[2-(2,6-dioxo-3-piperidyl)-1,3-dioxoisoindolin-4-yl]amino}propoxy)ethoxy]ethoxy}propyl)terephthalamide

 , ,
, , 
Abstract
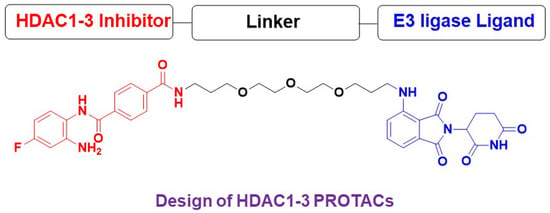
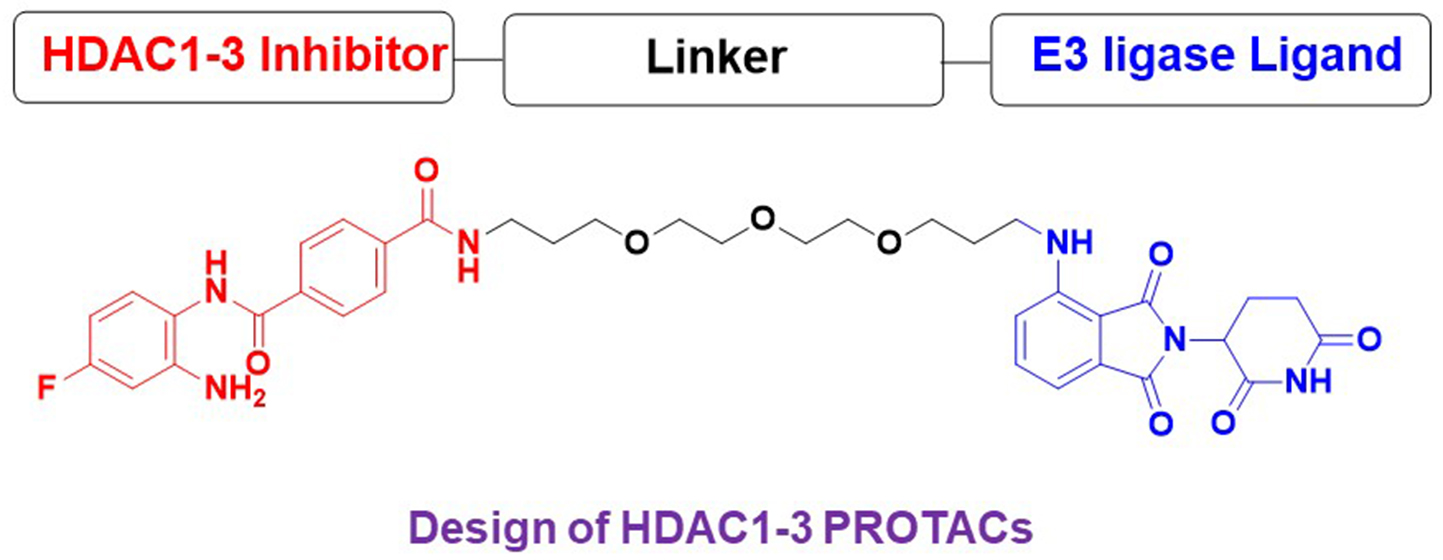
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In Vitro HDAC Inhibition Assay
2.2.2. Cellular Testing
3. Materials and Methods
3.1. General Experimental Information
3.2. Experimental Procedures and Characterization of Synthesized Compounds
3.3. Biological Testing
In Vitro HDAC Inhibition Assay
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pant, K.; Peixoto, E.; Richard, S.; Gradilone, S.A. Role of histone deacetylases in carcinogenesis: Potential role in cholangiocarcinoma. Cells 2020, 9, 780. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Abel, T.; Zukin, R.S. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr. Opin. Pharmacol. 2008, 8, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Miao, X.; Liu, Y.; Li, F.; Liu, Q.; Sun, J.; Cai, L. Dysregulation of Histone Acetyltransferases and Deacetylases in Cardiovascular Diseases. Oxid. Med. Cell. Longev. 2014, 2014, 641979. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Tsoi, L.C.; Gudjonsson, J.E. Dysregulated epigenetic modifications in psoriasis. Exp. Dermatol. 2021, 30, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; Jiang, Q.; Zhang, L.; Song, W. Zinc binding groups for histone deacetylase inhibitors. J. Enzym. Inhib. Med. Chem. 2018, 33, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.F.; Weïwer, M.; Lewis, M.C.; Holson, E.B. Small molecule inhibitors of zinc-dependent histone deacetylases. Neurotherapeutics 2013, 10, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.S.; Chan, A.H.Y.; Ganesan, A. Thirty Years of HDAC Inhibitors: 2020 Insight and Hindsight. J. Med. Chem. 2020, 63, 12460–12484. [Google Scholar] [CrossRef] [PubMed]
- Perrin, J.; Werner, T.; Kurzawa, N.; Rutkowska, A.; Childs, D.D.; Kalxdorf, M.; Poeckel, D.; Stonehouse, E.; Strohmer, K.; Heller, B. Identifying drug targets in tissues and whole blood with thermal-shift profiling. Nat. Biotechnol. 2020, 38, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Becher, I.; Werner, T.; Doce, C.; Zaal, E.A.; Tögel, I.; Khan, C.A.; Rueger, A.; Muelbaier, M.; Salzer, E.; Berkers, C.R. Thermal profiling reveals phenylalanine hydroxylase as an off-target of panobinostat. Nat. Chem. Biol. 2016, 12, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.S.; Abdelsalam, M.; Zeyn, Y.; Zessin, M.; Mustafa, A.-H.M.; Fischer, M.A.; Zeyen, P.; Sun, P.; Bülbül, E.F.; Vecchio, A.; et al. Synthesis, Molecular Docking and Biological Characterization of Pyrazine Linked 2-Aminobenzamides as New Class I Selective Histone Deacetylase (HDAC) Inhibitors with Anti-Leukemic Activity. Int. J. Mol. Sci. 2022, 23, 369. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Tang, P.; Yuan, Q. Rational design and characterization of a DNA/HDAC dual-targeting inhibitor containing nitrogen mustard and 2-aminobenzamide moieties. Medchemcomm 2018, 9, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.F.; Lundh, M.; Kaya, T.; McCarren, P.; Zhang, Y.-L.; Chattopadhyay, S.; Gale, J.P.; Galbo, T.; Fisher, S.L.; Meier, B.C.; et al. An Isochemogenic Set of Inhibitors to Define the Therapeutic Potential of Histone Deacetylases in β-Cell Protection. ACS Chem. Biol. 2016, 11, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef] [PubMed]
- Bricelj, A.; Steinebach, C.; Kuchta, R.; Gütschow, M.; Sosič, I. E3 Ligase Ligands in Successful PROTACs: An Overview of Syntheses and Linker Attachment Points. Front. Chem. 2021, 9, 707317. [Google Scholar] [CrossRef] [PubMed]
- Schiedel, M.; Herp, D.; Hammelmann, S.R.; Swyter, S.R.; Lehotzky, A.; Robaa, D.; Oláh, J.; Ovadi, J.; Sippl, W.; Jung, M. Chemically induced degradation of sirtuin 2 (Sirt2) by a proteolysis targeting chimera (PROTAC) based on sirtuin rearranging ligands (SirReals). J. Med. Chem. 2018, 61, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Darwish, S.; Ghazy, E.; Heimburg, T.; Herp, D.; Zeyen, P.; Salem-Altintas, R.; Ridinger, J.; Robaa, D.; Schmidtkunz, K.; Erdmann, F.; et al. Design, Synthesis and Biological Characterization of Histone Deacetylase 8 (HDAC8) Proteolysis Targeting Chimeras (PROTACs) with Anti-Neuroblastoma Activity. Int. J. Mol. Sci. 2022, 23, 7535. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Li, Y.; Wang, X.; Dong, G.; Sheng, C. Discovery of Novel PDEδ Degraders for the Treatment of KRAS Mutant Colorectal Cancer. J. Med. Chem. 2020, 63, 7892–7905. [Google Scholar] [CrossRef] [PubMed]
- Zessin, M.; Kutil, Z.F.; Meleshin, M.; Nováková, Z.; Ghazy, E.; Kalbas, D.; Marek, M.; Romier, C.; Sippl, W.; Bařinka, C. One-atom substitution enables direct and continuous monitoring of histone deacylase activity. Biochemistry 2019, 58, 4777–4789. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Cpd. No. | Structure | HDAC1 (IC50 µM) | HDAC2 (IC50 µM) | HDAC3 (IC50 µM) |
|---|---|---|---|---|
| 5 |  | 18.0 ± 1 | 14.0 ± 1 | 3.4 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelsalam, M.; Zessin, M.; Schmidt, M.; Schutkowski, M.; Sippl, W. N4-(2-Amino-4-fluorophenyl)-N1-(3-{2-[2-(3-{[2-(2,6-dioxo-3-piperidyl)-1,3-dioxoisoindolin-4-yl]amino}propoxy)ethoxy]ethoxy}propyl)terephthalamide. Molbank 2022, 2022, M1501. https://doi.org/10.3390/M1501
Abdelsalam M, Zessin M, Schmidt M, Schutkowski M, Sippl W. N4-(2-Amino-4-fluorophenyl)-N1-(3-{2-[2-(3-{[2-(2,6-dioxo-3-piperidyl)-1,3-dioxoisoindolin-4-yl]amino}propoxy)ethoxy]ethoxy}propyl)terephthalamide. Molbank. 2022; 2022(4):M1501. https://doi.org/10.3390/M1501
Chicago/Turabian StyleAbdelsalam, Mohamed, Matthes Zessin, Matthias Schmidt, Mike Schutkowski, and Wolfgang Sippl. 2022. "N4-(2-Amino-4-fluorophenyl)-N1-(3-{2-[2-(3-{[2-(2,6-dioxo-3-piperidyl)-1,3-dioxoisoindolin-4-yl]amino}propoxy)ethoxy]ethoxy}propyl)terephthalamide" Molbank 2022, no. 4: M1501. https://doi.org/10.3390/M1501
APA StyleAbdelsalam, M., Zessin, M., Schmidt, M., Schutkowski, M., & Sippl, W. (2022). N4-(2-Amino-4-fluorophenyl)-N1-(3-{2-[2-(3-{[2-(2,6-dioxo-3-piperidyl)-1,3-dioxoisoindolin-4-yl]amino}propoxy)ethoxy]ethoxy}propyl)terephthalamide. Molbank, 2022(4), M1501. https://doi.org/10.3390/M1501