
3-Methyl 5-{3-[(4-Methylbenzenesulfonyl)oxy]propyl} 4-(2,3-Dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate



Abstract
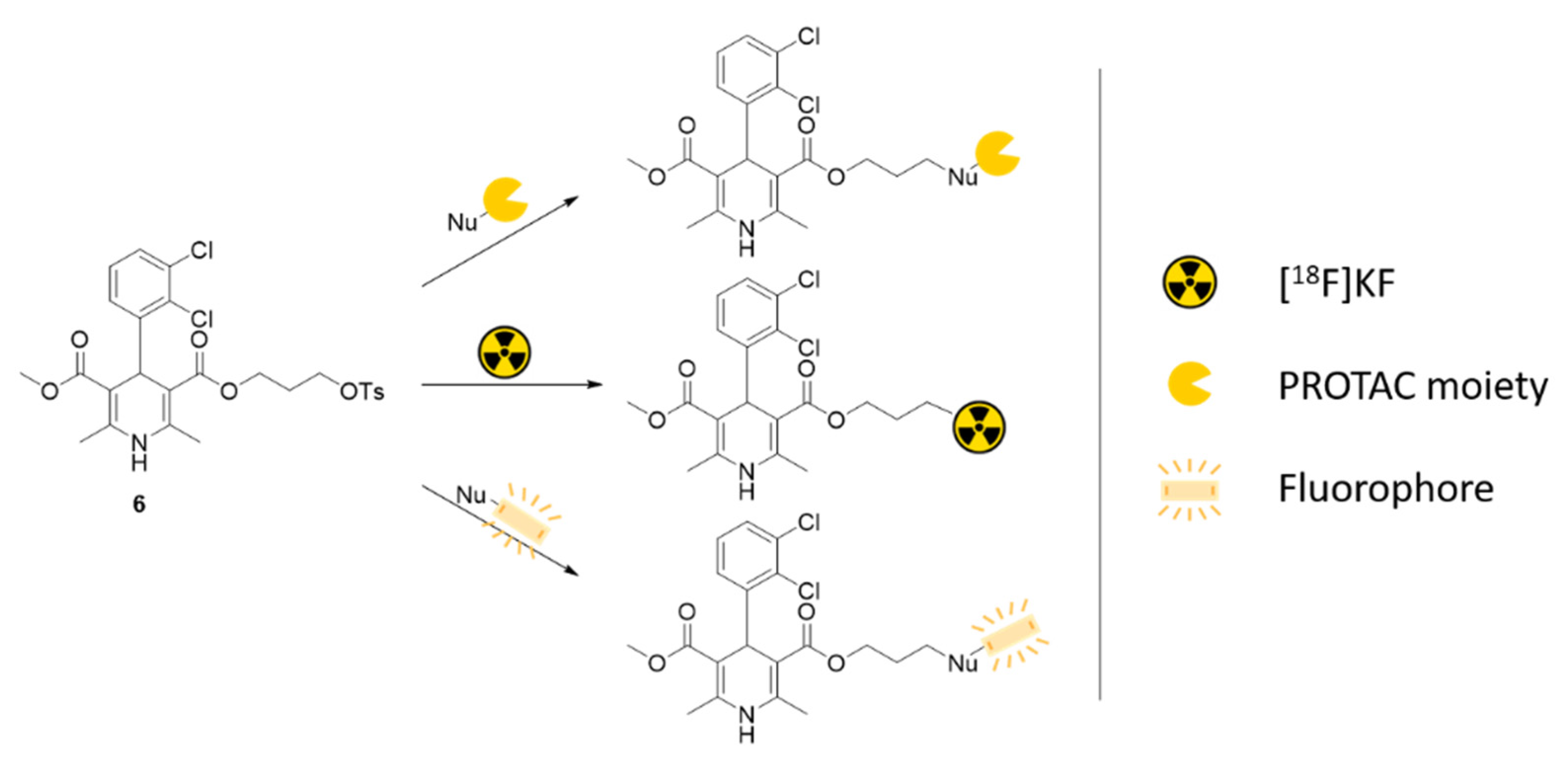
1. Introduction
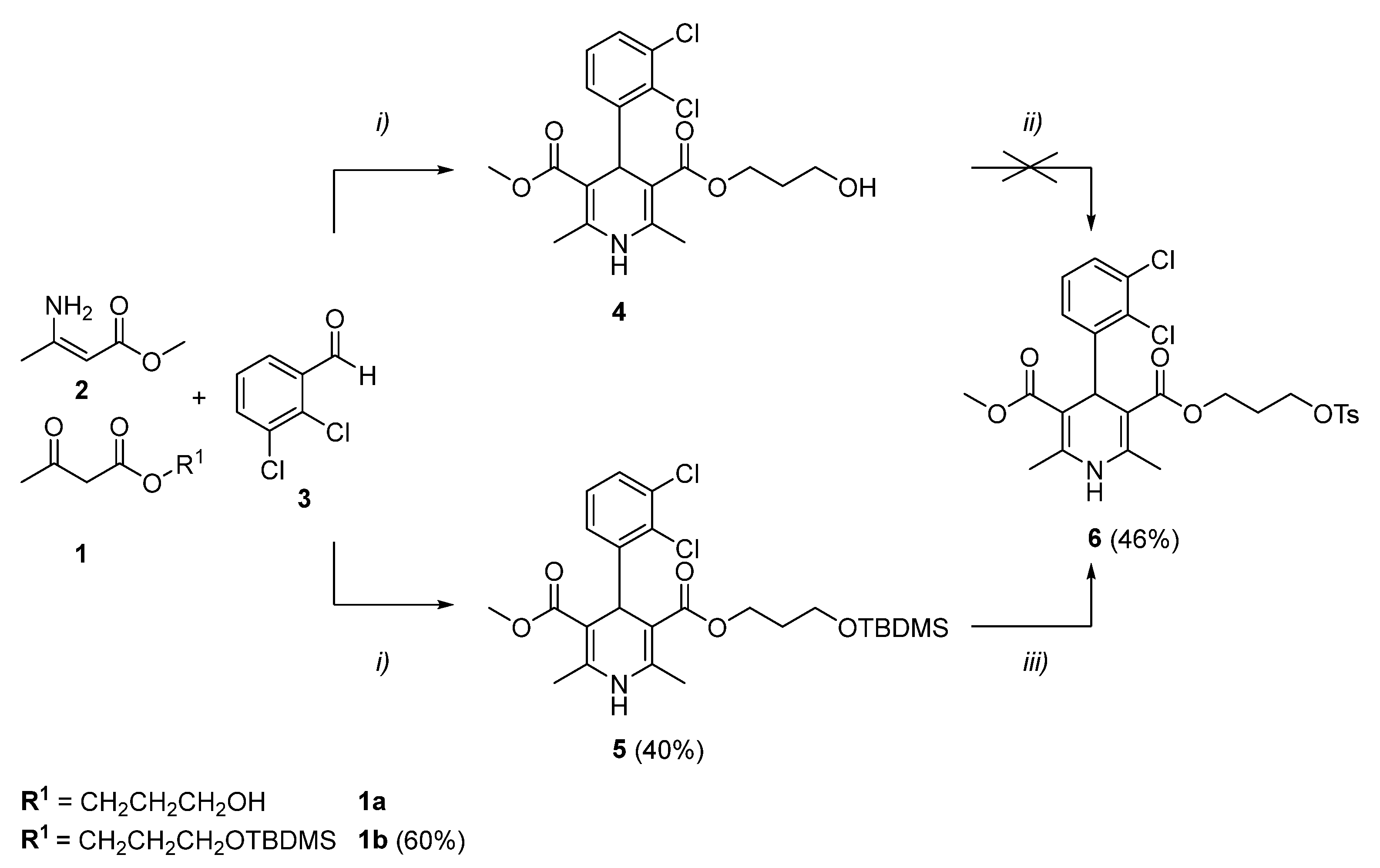
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Information
3.2. Experimental Procedures and Characterization
- 3-[(tert-butyldimethylsilyl)oxy]propan-1-ol
- 3-[(tert-butyldimethylsilyl)oxy]propyl 3-oxobutanoate (1b)
- 3-{3-[(tert-butyldimethylsilyl)oxy]propyl} 5-methyl 4-(2,3-dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (5)
- 3-methyl 5-{3-[(4-methylbenzenesulfonyl)oxy]propyl} 4-(2,3-dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate (6)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hantzsch, A. Condensationsprodukte aus Aldehydammoniak und ketonartigen Verbindungen. Ber. Dtsch. Chem. Ges. 1881, 14, 1637–1638. [Google Scholar] [CrossRef]
- Auria-Luna, F.; Marqués-López, E.; Herrera, R.P. Organocatalytic Enantioselective Synthesis of 1,4-Dihydropyridines. Adv. Synth. Catal. 2017, 359, 2161–2175. [Google Scholar] [CrossRef]
- Das Sarma, M.; Ghosh, S. Recent Advances in Catalysis of Hantzsch and Related Synthesis of 1,4-Dihydropyridines and Polyhydroquinolines: A Brief Overview. Asian J. Chem. 2020, 32, 2943–2952. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V. Construction and Aromatization of Hantzsch 1,4-Dihydropyridines under Microwave Irradiation: A Green Approach. ChemistrySelect 2022, 7, e202104032. [Google Scholar] [CrossRef]
- Fares, H.; DiNicolantonio, J.J.; Keefe, J.H.; Lavie, C.J. Amlodipine in hypertension: A first-line agent with efficacy for improving blood pressure and patient outcomes. Open Heart 2016, 3, e000473. [Google Scholar] [CrossRef]
- Johnson, B.A.; Ait-Daoud, N.; Wells, L.T. Effects of Isradipine, a Dihydropyridine-Class Calcium Channel Antagonist, on D-Methamphetamine-Induced Cognitive and Physiological Changes in Humans. Neuropsychopharmacology 2000, 22, 504–512. [Google Scholar] [CrossRef]
- Mancia, G.; Parati, G.; Bilo, G.; Choi, J.; Kilama, M.O.; Ruilope, L.M.; TALENT Investigators. Blood pressure control by the nifedipine GITS–telmisartan combination in patients at high cardiovascular risk: The TALENT study. J. Hypertens. 2011, 29, 600–609. [Google Scholar] [CrossRef]
- Toal, C.B.; Meredith, P.A.; Elliott, H.L. Long-acting dihydropyridine calcium-channel blockers and sympathetic nervous system activity in hypertension: A literature review comparing amlodipine and nifedipine GITS. Blood Press. 2012, 21, 3–10. [Google Scholar] [CrossRef]
- Wang, J.-G.; Kario, K.; Lau, T.; Wei, Y.Q.; Park, C.G.; Kim, C.H.; Huang, J.; Zhang, W.; Li, Y.; Yan, P.; et al. Use of dihydropyridine calcium channel blockers in the management of hypertension in Eastern Asians: A scientific statement from the Asian Pacific Heart Association. Hypertens. Res. 2011, 34, 423–430. [Google Scholar] [CrossRef]
- Goto, R.N.; Sobral, L.M.; Sousa, L.O.; Garcia, C.B.; Lopes, N.P.; Marín-Prida, J.; Ochoa-Rodríguez, E.; Verdecia-Reyes, Y.; Pardo-Andreu, G.L.; Curti, C.; et al. Anti-cancer activity of a new dihydropyridine derivative, VdiE-2N, in head and neck squamous cell carcinoma. Eur. J. Pharmacol. 2018, 819, 198–206. [Google Scholar] [CrossRef]
- Ošiņa, K.; Rostoka, E.; Isajevs, S.; Sokolovska, J.; Sjakste, T.; Sjakste, N. Effects of an Antimutagenic 1,4-Dihydropyridine AV-153 on Expression of Nitric Oxide Synthases and DNA Repair-related Enzymes and Genes in Kidneys of Rats with a Streptozotocin Model of Diabetes Mellitus. Basic Clin. Pharmacol. Toxicol. 2016, 119, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, R.; Maheswari, S.; Murugesan, S.; Sandhiya, R.; Lalitha, A. Catalyst-free one-pot synthesis and antioxidant evaluation of highly functionalized novel 1,4-dihydropyridine derivatives. Res. Chem. Intermed. 2015, 41, 8233–8243. [Google Scholar] [CrossRef]
- Michalska, P.; Mayo, P.; Fernández-Mendívil, C.; Tenti, G.; Duarte, P.; Buendia, I.; Ramos, M.T.; López, M.G.; Menéndez, J.C.; León, R. Antioxidant, Anti-inflammatory and Neuroprotective Profiles of Novel 1,4-Dihydropyridine Derivatives for the Treatment of Alzheimer’s Disease. Antioxidants 2020, 9, 650. [Google Scholar] [CrossRef] [PubMed]
- Malhi, D.S.; Kaur, M.; Sohal, H.S. Effect of Substitutions on 1,4-Dihdropyridines to Achieve Potential Anti-Microbial Drugs: A Review. ChemistrySelect 2019, 4, 11321–11336. [Google Scholar] [CrossRef]
- Baumert, C.; Günthel, M.; Krawczyk, S.; Hemmer, M.; Wersig, T.; Langner, A.; Molnár, J.; Lage, H.; Hilgeroth, A. Development of small-molecule P-gp inhibitors of the N-benzyl 1,4-dihydropyridine type: Novel aspects in SAR and bioanalytical evaluation of multidrug resistance (MDR) reversal properties. Bioorg. Med. Chem. 2013, 21, 166–177. [Google Scholar] [CrossRef]
- Khoshneviszadeh, M.; Edraki, N.; Javidnia, K.; Alborzi, A.; Pourabbas, B.; Mardaneh, J.; Miri, R. Synthesis and biological evaluation of some new 1,4-dihydropyridines containing different ester substitute and diethyl carbamoyl group as anti-tubercular agents. Bioorg. Med. Chem. 2009, 17, 1579–1586. [Google Scholar] [CrossRef]
- Längle, D.; Marquardt, V.; Heider, E.; Vigante, B.; Duburs, G.; Luntena, I.; Flötgen, D.; Golz, C.; Strohmann, C.; Koch, O.; et al. Design, synthesis and 3D-QSAR studies of novel 1,4-dihydropyridines as TGFβ/Smad inhibitors. Eur. J. Med. Chem. 2015, 95, 249–266. [Google Scholar] [CrossRef]
- Pollo, L.A.E.; de Moraes, M.H.; Cisilotto, J.; Creczynski-Pasa, T.B.; Biavatti, M.W.; Steindel, M.; Sandjo, L.P. Synthesis and in vitro evaluation of Ca2+ channel blockers 1,4-dihydropyridines analogues against Trypanosoma cruzi and Leishmania amazonensis: SAR analysis. Parasitol. Int. 2017, 66, 789–797. [Google Scholar] [CrossRef]
- Yamamoto, T.; Niwa, S.; Ohno, S.; Tokumasu, M.; Masuzawa, Y.; Nakanishi, C.; Nakajo, A.; Onishi, T.; Koganei, H.; Fujita, S.-I.; et al. The structure–activity relationship study on 2-, 5-, and 6-position of the water soluble 1,4-dihydropyridine derivatives blocking N-type calcium channels. Bioorg. Med. Chem. Lett. 2008, 18, 4813–4816. [Google Scholar] [CrossRef]
- Ling, Y.; Hao, Z.Y.; Liang, D.; Zhang, C.L.; Liu, Y.F.; Wang, Y. The Expanding Role of Pyridine and Dihydropyridine Scaffolds in Drug Design. Drug Des. Devel. Ther. 2021, 15, 4289–4338. [Google Scholar] [CrossRef]
- Sepehri, S.; Sanchez, H.P.; Fassihi, A. Hantzsch-Type Dihydropyridines and Biginelli-Type Tetra-hydropyrimidines: A Review of their Chemotherapeutic Activities. J. Pharm. Pharm. Sci. 2015, 18, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Pleiss, U. 1,4-Dihydropyridines (DHPs)—A class of very potent drugs: Syntheses of isotopically labeled DHP derivatives during the last four decades. J. Label. Compd. Radiopharm. 2007, 50, 818–830. [Google Scholar] [CrossRef]
- Sadeghpour, H.; Jalilian, A.R.; Akhlaghi, M.; Shafiee, A.; Mirzaii, M.; Miri, R.; Saddadi, F. Biological evaluation of F-18 -nifedipine as a novel PET tracer for L-type calcium channel imaging. Nukleonika 2008, 53, 151–154. [Google Scholar]
- Ding, R.; He, Y.; Wang, X.; Xu, J.; Chen, Y.; Feng, M.; Qi, C. Treatment of alcohols with tosyl chloride does not always lead to the formation of tosylates. Molecules 2011, 16, 5665–5673. [Google Scholar] [CrossRef] [PubMed]
- Kabalka, G.W.; Varma, M.; Varma, R.S.; Srivastava, P.C.; Knapp, F.F. TOSYLATION OF ALCOHOLS. J. Org. Chem. 1986, 51, 2386–2388. [Google Scholar] [CrossRef]
- Lambert, C.; Mease, R.C.; Avren, L.; Le, T.; Sabet, H.; McAfee, J.G. Radioiodinated (aminostyryl)pyridinium (ASP) dyes: New cell membrane probes for labeling mixed leukocytes and lymphocytes for diagnostic imaging. Nucl. Med. Biol. 1996, 23, 417–427. [Google Scholar] [CrossRef]
- Gembus, V.; Marsais, F.; Levacher, V. An Efficient Organocatalyzed Interconversion of Silyl Ethers to Tosylates Using DBU and p-Toluenesulfonyl Fluoride. Synlett 2008, 2008, 1463–1466. [Google Scholar] [CrossRef]
- Clemens, R.J.; Hyatt, J.A. Acetoacetylation with 2,2,6-trimethyl-4H-1,3-dioxin-4-one: A convenient alternative to diketene. J. Org. Chem. 1985, 50, 2431–2435. [Google Scholar] [CrossRef]
- Lagoutte, R.; Pastor, M.; Berthet, M.; Winssinger, N. Rapid and scalable synthesis of chiral bromolactones as precursors to α-exo-methylene-γ-butyrolactone-containing sesquiterpene lactones. Tetrahedron 2018, 74, 6012–6021. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}

| Entry | Base (Equiv.) | Time (h) | T (°C) | Yield (%) |
|---|---|---|---|---|
| 1 | DBU 0.2 | 24 | 25 | 8 |
| 2 | DBU 0.2 | 24 | 70 | - |
| 3 | TBAF 0.2 | 48 | 25 | - * |
| 4 | TBAF 1 | 48 | 25 | - * |
| 5 | DBU 0.6 | 24 | 25 | 46 |
| 6 | DBU 1 | 24 | 25 | 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borgarelli, C.; Lenaers, S.; De Borggraeve, W.M.; Ismalaj, E. 3-Methyl 5-{3-[(4-Methylbenzenesulfonyl)oxy]propyl} 4-(2,3-Dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate. Molbank 2022, 2022, M1460. https://doi.org/10.3390/M1460
Borgarelli C, Lenaers S, De Borggraeve WM, Ismalaj E. 3-Methyl 5-{3-[(4-Methylbenzenesulfonyl)oxy]propyl} 4-(2,3-Dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate. Molbank. 2022; 2022(4):M1460. https://doi.org/10.3390/M1460
Chicago/Turabian StyleBorgarelli, Carlotta, Stijn Lenaers, Wim M. De Borggraeve, and Ermal Ismalaj. 2022. "3-Methyl 5-{3-[(4-Methylbenzenesulfonyl)oxy]propyl} 4-(2,3-Dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate" Molbank 2022, no. 4: M1460. https://doi.org/10.3390/M1460
APA StyleBorgarelli, C., Lenaers, S., De Borggraeve, W. M., & Ismalaj, E. (2022). 3-Methyl 5-{3-[(4-Methylbenzenesulfonyl)oxy]propyl} 4-(2,3-Dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate. Molbank, 2022(4), M1460. https://doi.org/10.3390/M1460