
2,3,5-Tri-O-benzyl-d-xylofuranose




Abstract
1. Introduction
2. Results and Discussion
2.1. Synthesis
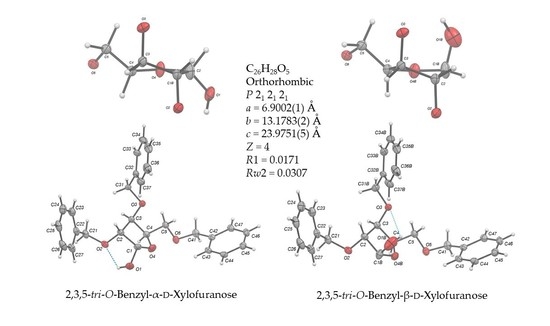
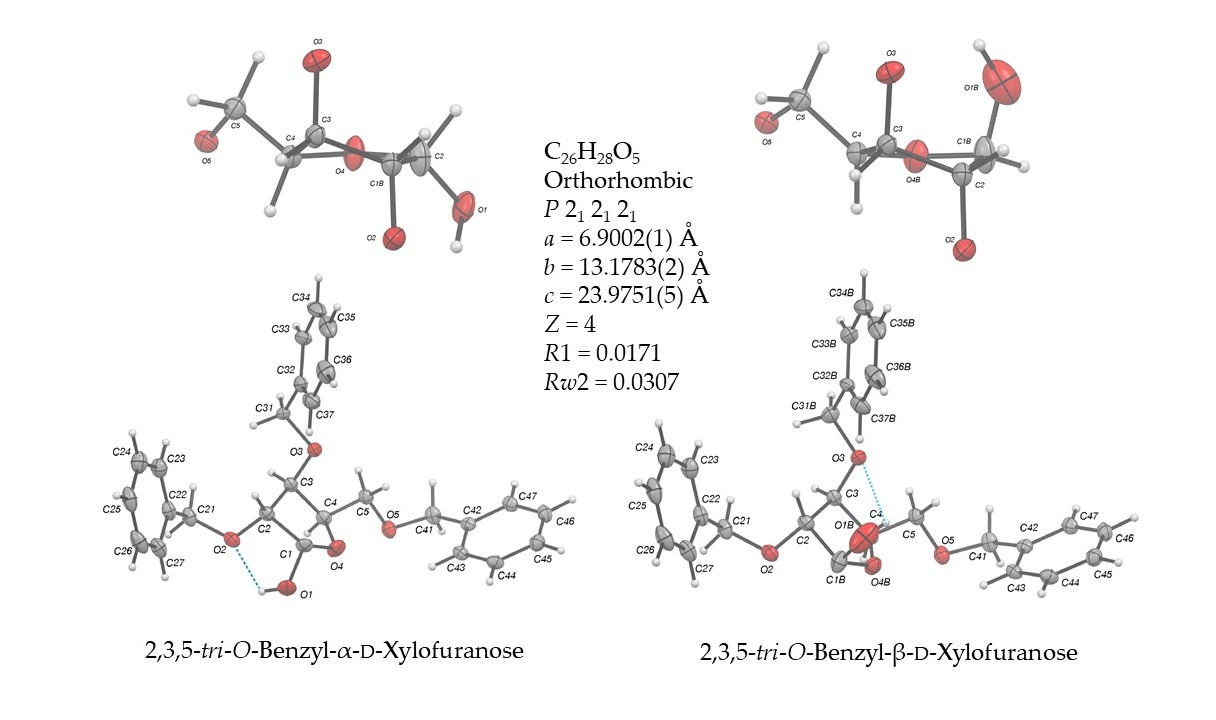
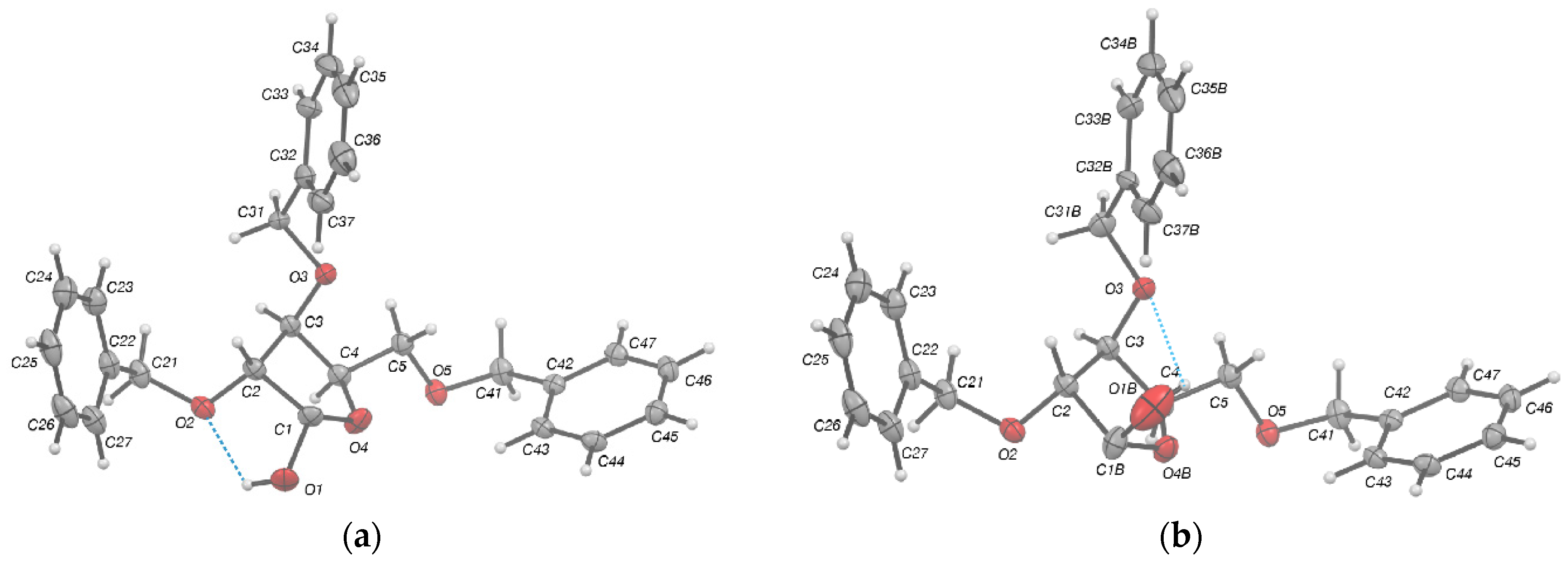
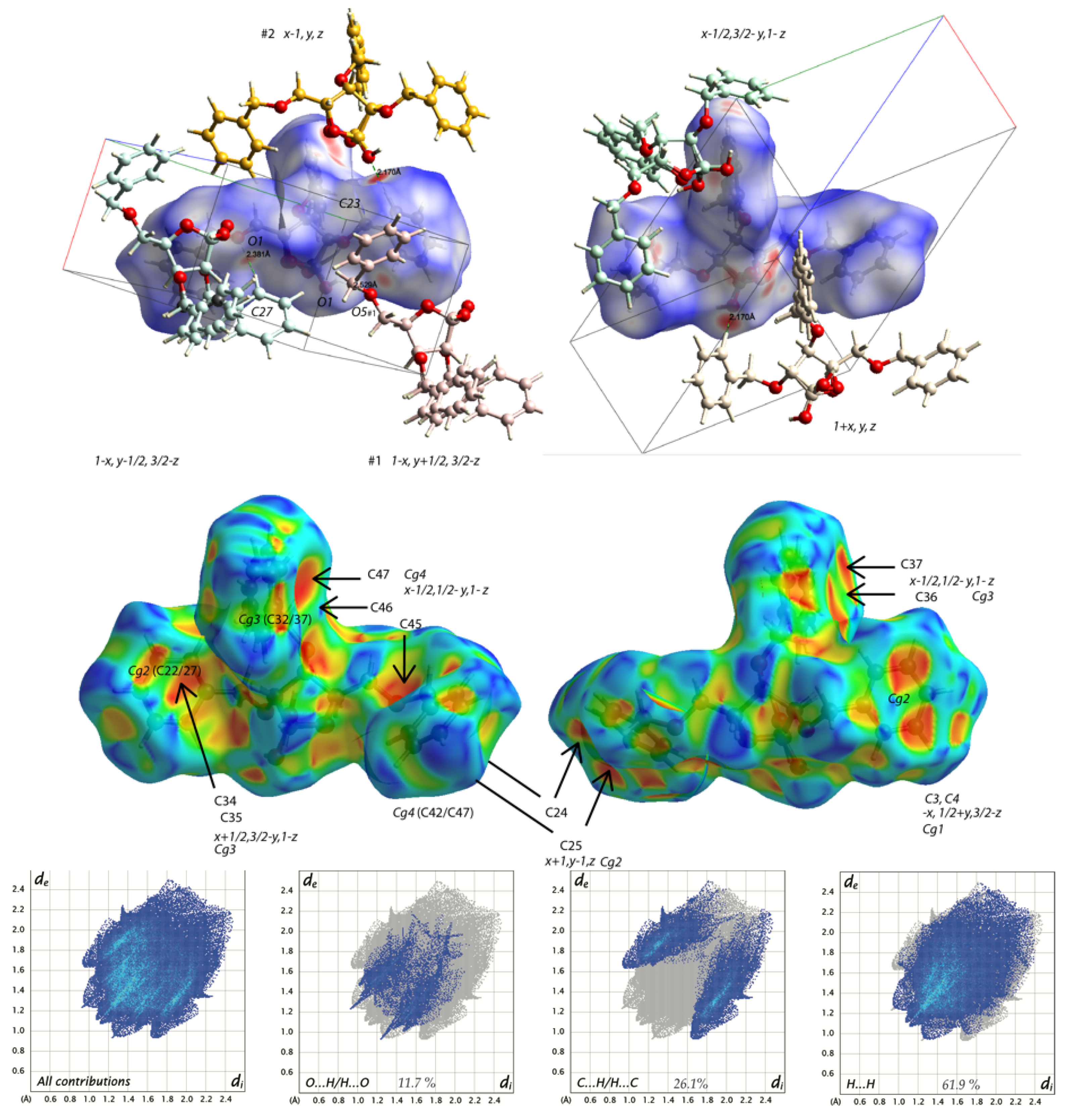
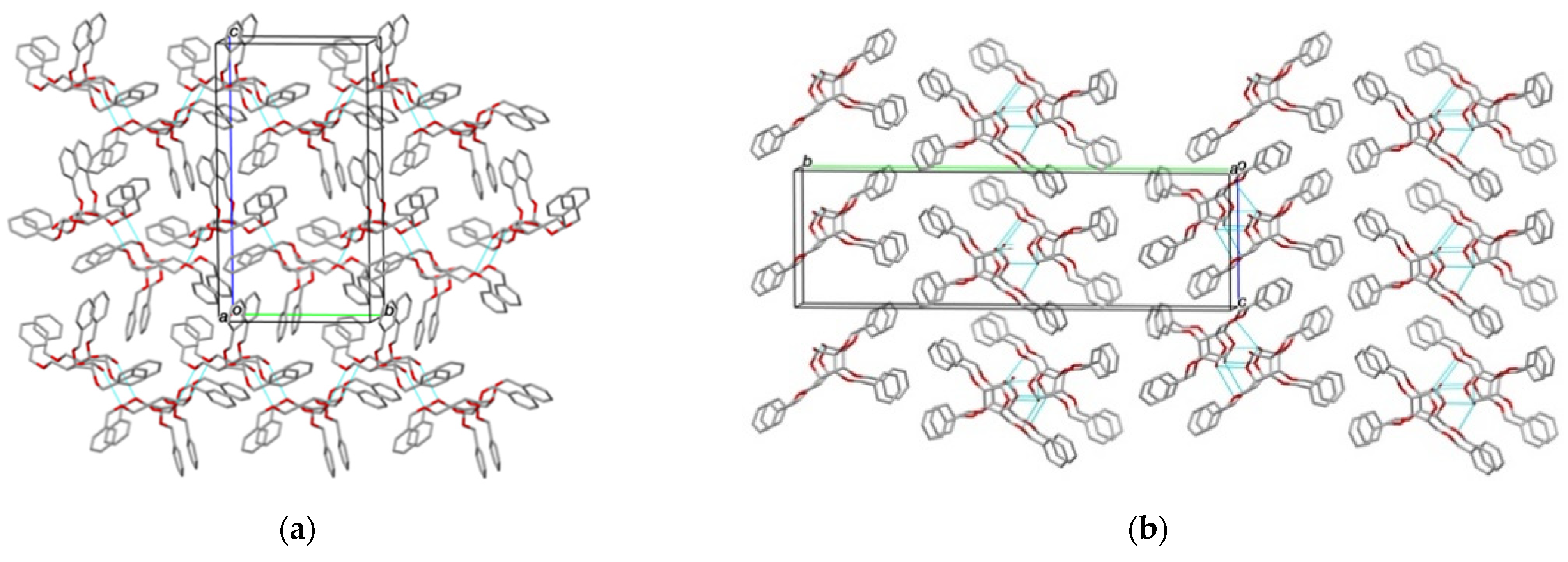
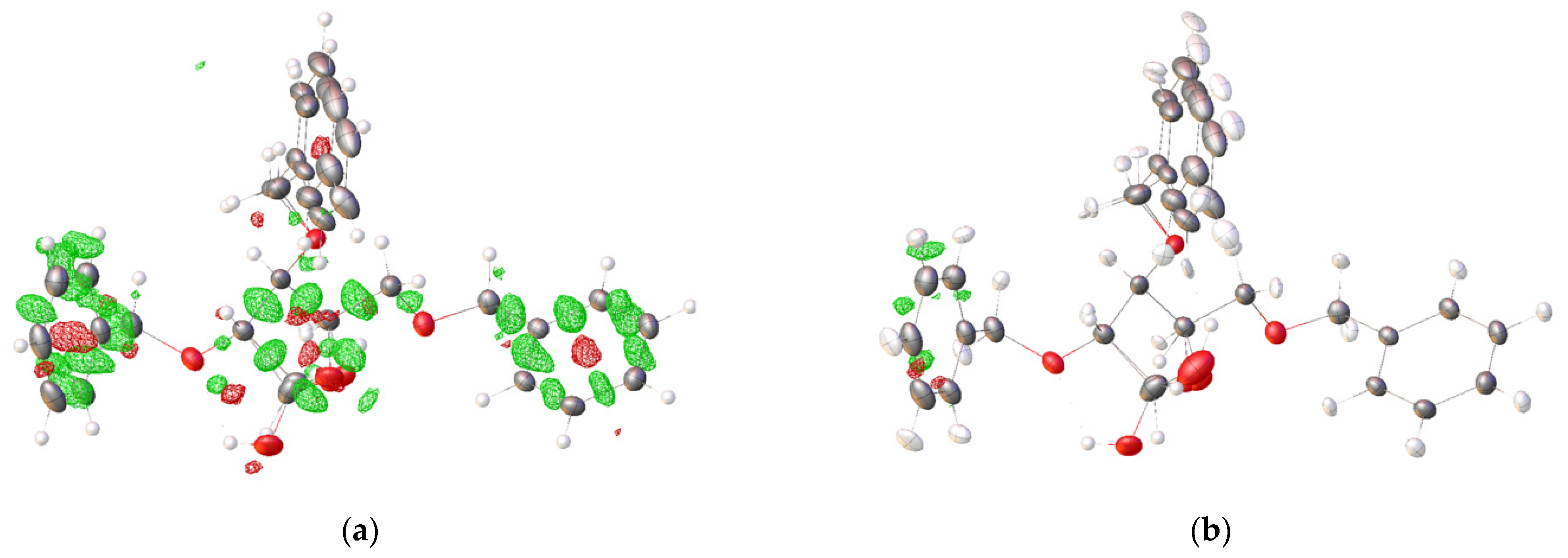
2.2. Structural Commentary
3. Materials and Methods
3.1. General Remarks
3.2. Procedures and Characterization Data
3.2.1. Synthesis of 2,3,5-Tri-O-benzyl-α,β-d-xylofuranose (α,β)-1
3.2.2. Crystallization of 2,3,5-Tri-O-benzyl-α,β-d-xylofuranose (α,β)-1 (ca. 89:11)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ernst, B.; Hart, G.W.; Sinaÿ, P. (Eds.) Carbohydrates in Chemistry and Biology; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Tiwari, V.K. (Ed.) Carbohydrates in Drug Discovery and Development: Synthesis and Application, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2020; ISBN 9780128166758. [Google Scholar]
- Tamburrini, A.; Colombo, C.; Bernardi, A. Design and synthesis of glycomimetics: Recent advances. Med. Res. Rev. 2020, 40, 495–531. [Google Scholar] [CrossRef] [PubMed]
- Sattin, S.; Bernardi, A. Design and synthesis of glycomimetics. In Carbohydrate Chemistry; Rauter, A.P., Lindhorst, T.K., Queneau, Y., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2016; Volume 41, pp. 1–25. ISBN 978-1-78262-121-8. [Google Scholar]
- Ernst, B.; Magnani, J.L. From carbohydrate leads to glycomimetic drugs. Nat. Rev. Drug Discov. 2009, 8, 661–677. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Martin, O.R. (Eds.) Iminosugars: From Synthesis to Therapeutic Applications; John Wiley & Sons: Chichester, UK, 2007; ISBN 978-0-470-03391-3. [Google Scholar]
- Asano, N. Iminosugars: The Potential of Carbohydrate Analogs. In Carbohydrate Chemistry: State of the Art and Challenges for Drug Development; Cipolla, L., Ed.; Imperial College Press: London, UK, 2016; pp. 279–301. ISBN 978-1783267194. [Google Scholar]
- Harit, V.K.; Ramesh, N.G. Amino-functionalized iminocyclitols: Synthetic glycomimetics of medicinal interest. RSC Adv. 2016, 6, 109528–109607. [Google Scholar] [CrossRef]
- Conforti, I.; Marra, A. Iminosugars as glycosyltransferase inhibitors. Org. Biomol. Chem. 2021, 19, 5439–5475. [Google Scholar] [CrossRef] [PubMed]
- Alonzi, D.S.; Scott, K.A.; Dwek, R.A.; Zitzmann, N. Iminosugar antivirals: The therapeutic sweet spot. Biochem. Soc. Trans. 2017, 45, 571–582. [Google Scholar] [CrossRef]
- Horne, G. Alkaloids as important scaffolds in therapeutic drugs for the treatments of cancer, tuberculosis, and smoking cessation. Top. Med. Chem. 2014, 12, 23–51. [Google Scholar] [CrossRef]
- Nicolas, C.; Engo-Ilanga, F.; Cocaud, C.; Martin, O.R. En Route to Novel Furanoside Mimics Through Stereoselective Zinc-Mediated Propargylation of N-Benzyl-glycofuranosylamines Using Ultrasound Activation. Synlett 2015, 26, 187–192. [Google Scholar] [CrossRef]
- Cocaud, C.; Maujoin, A.; Zheng, R.B.; Lowary, T.L.; Rodrigues, N.; Percina, N.; Chartier, A.; Buron, F.; Routier, S.; Nicolas, C.; et al. Triazole-Linked Iminosugars and Aromatic Systems as Simplified UDP-Galf Mimics: Synthesis and Preliminary Evaluation as Galf-transferase Inhibitors. Eur. J. Org. Chem. 2017, 41, 6192–6201. [Google Scholar] [CrossRef]
- Cocaud, C.; Nicolas, C.; Poisson, T.; Pannecoucke, X.; Legault, C.Y.; Martin, O.R. Tunable Approach for the Stereoselective Synthesis of 1-C-Diethylphosphono(difluoromethyl) Iminosugars as Glycosyl Phosphate Mimics. J. Org. Chem. 2017, 82, 2753–2763. [Google Scholar] [CrossRef]
- Cocaud, C.; Zheng, R.B.; Lowary, T.; Poisson, T.; Pannecoucke, X.; Nicolas, C.; Martin, O.R. 1-C-Phosphonomethyl- and 1-C-Difluorophosphonomethyl-1,4-imino-l-arabinitols as Galf Transferase Inhibitors: A Comparison. Carbohydr. Res. 2018, 461, 45–50. [Google Scholar] [CrossRef]
- Nicolas, C.; Martin, O.R. Glycoside Mimics From Glycosylamines: Recent Progress. Molecules 2018, 23, 1612. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jaszczyk, J.; Pannecoucke, X.; Poisson, T.; Martin, O.R.; Nicolas, C. Stereospecific Synthesis of Glycoside Mimics Through Migita–Kosugi–Stille Cross-Coupling Reactions of Chemically and Configurationally Stable 1-C-Tributylstannyl Iminosugars. Adv. Synth. Catal. 2021, 363, 470–483. [Google Scholar] [CrossRef]
- Chronowska, A.; Gallienne, E.; Nicolas, C.; Kato, A.; Adachi, I.; Martin, O.R. An expeditious synthesis of an analogue of (−)-steviamine by way of the 1,3-dipolar cycloaddition of a nitrile oxide with a 1-C-allyl iminosugar. Tetrahedron Lett. 2011, 52, 6399–6402. [Google Scholar] [CrossRef]
- Ernholt, B.V.; Thomsen, I.B.; Lohse, A.; Plesner, I.W.; Jensen, K.B.; Hazell, R.G.; Liang, X.; Jakobsen, A.; Bols, M. Enantiospecific Synthesis of 1-Azafagomine. Chem. Eur. J. 2000, 6, 278–287. [Google Scholar] [CrossRef]
- Yu, C.-Y.; Huang, M.-H. Radicamines A and B: Synthesis and Revision of the Absolute Configuration. Org. Lett. 2006, 8, 3021–3024. [Google Scholar] [CrossRef]
- Liu, C.; Gao, J.; Yang, G.; Wightman, R.H.; Jiang, S. Enantiospecific Synthesis of (–)-Radicamine B. Lett. Org. Chem. 2007, 4, 556–558. [Google Scholar] [CrossRef][Green Version]
- Van Rijssel, E.R.; van Delft, P.; Lodder, G.; Overkleeft, H.S.; van der Marel, G.A.; Filippov, D.V.; Codée, J.D.C. Furanosyl Oxocarbenium Ion Stability and Stereoselectivity. Angew. Chem. Int. Ed. 2014, 53, 10381–10385. [Google Scholar] [CrossRef] [PubMed]
- Wennekes, T.; Bonger, K.M.; Vogel, K.; van den Berg, R.J.B.H.N.; Strijland, A.; Donker-Koopman, W.E.; Aerts, J.M.F.G.; van der Marel, G.A.; Overkleeft, H.S. The Development of an Aza-C-Glycoside Library Based on a TandemStaudinger/Aza-Wittig/Ugi Three-Component Reaction. Eur. J. Org. Chem. 2012, 32, 6420–6454. [Google Scholar] [CrossRef]
- Evangelista, T.C.S.; Lopéz, Ó.; Sydnes, M.O.; Fernández-Bolaños, J.G.; Ferreira, S.B.; Lindbäck, E. Bicyclic 1-Azafagomine Derivatives: Synthesis and Glycosidase Inhibitory Testing. Synthesis 2019, 51, 4066–4077. [Google Scholar] [CrossRef]
- Altona, C.; Sundaralingam, M. Conformational Analysis of the Sugar Ring in Nucleosides and Nucleotides. A New Description Using the Concept of Pseudorotation. J. Am. Chem. Soc. 1972, 94, 8205–8212. [Google Scholar] [CrossRef]
- Taha, H.A.; Richards, M.R.; Lowary, T.L. Conformational Analysis of Furanoside-Containing Mono- and Oligosaccharides. Chem. Rev. 2013, 113, 1851–1876. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Suthagar, K.; Polson, M.I.J.; Fairbanks, A.J. Unexpected furanose/pyranose equilibration of N-glycosyl sulfonamides, sulfamides and sulfamates. Org. Biomol. Chem. 2015, 13, 6573–6579. [Google Scholar] [CrossRef] [PubMed]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Rigaku, O.D. CrystalClear, version 2.0; Rigaku Corporation: Tokyo, Japan, 2008.
- Rigaku, O.D. CrysAlisPro, version 1.171.41.121a; Rigaku Oxford Diffraction: Yarnton, UK, 2021.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Fugel, M.; Jayatilaka, D.; Hupf, E.; Overgaard, J.; Hathwar, V.R.; Macchi, P.; Turner, M.J.; Howard, J.A.K.; Dolomanov, O.V.; Puschmann, H.; et al. Probing the accuracy and precision of Hirshfeld atom refinement with HARt interfaced with Olex2. IUCrJ 2018, 5, 32–44. [Google Scholar] [CrossRef]
- Kleemiss, F.; Dolomanov, O.V.; Bodensteiner, M.; Peyerimhoff, N.; Midgley, L.; Bourhis, L.J.; Genoni, A.; Malaspina, L.A.; Jayatilaka, D.; Spencer, J.L.; et al. Accurate crystal structures and chemical properties from NoSpherA2. Chem. Sci. 2021, 12, 1675–1692. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.; Flack, H.D.; Wagner, T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2013, 69, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D—H...A | d(D—H) | d(H...A) | d(D...A) | <(DHA) |
|---|---|---|---|---|
| O1—H1...O2 | 0.964(10) | 1.938(11) | 2.5739(7) | 121.3(8) |
| O1—H1...O5 #1 | 0.964(10) | 2.548(10) | 3.3601(7) | 142.0(8) |
| O1B—H1B...O3 | 0.964(10) | 2.299(11) | 2.9095(7) | 120.4(8) |
| C23—H23...O1 #2 | 1.010(7) | 2.299(11) | 3.1929(7) | 158.84(8) |
| C27—H27...O5 #1 | 1.084(8) 1 | 2.3889(11) 1 | 3.4537(7) | 166.88(8) |
| Torsion Angle | (α)-I | (β)-I | Molecule A (4) | Molecule B (4) |
|---|---|---|---|---|
| C4—O4—C1—C2 | 4.55 (11) | 17.4 (10) | 32.47 (3) | 33.58 (3) |
| O4—C1—C2—C3 | −24.39 (13) | −29.29 (12) | 41.02 (3) | 41.29 (3) |
| C1—C2—C3—C4 | 33.41 (7) | 32.07 (11) | 34.19 (3) | 33.51 (3) |
| C1—O4—C4—C3 | 17.55 (9) | 2.2 (6) | 15.85 (3) | 13.88 (3) |
| C2—C3—C4—O4 | −32.07 (10) | −21.6 (8) | 10.74 (3) | −13.07 (3) |
| H1—O1—C1—O4 | −99.8 (6) | 67.17 (8) | −54.2 (3) | −66.8 |
| Χ (1) | syn | syn | syn | syn |
| C3—C4—C5—O5 | −167.42 (5) | −167.42 (5) | −172.2 (3) | −173.8 |
| γt/γ+/γ− (2) | γt | γt | γt | γt |
| Phase angle (P) | 11.1 (2) | 13.5(17) | 327.3 | 324.4 |
| Puckering amplitude (νmax) | 35.0 (1) | 32.6 (7) | 41.3 | 41.6 |
| S or N (3) | N | N | N | N |
| Identification code | 2,3,5-Tri-O-benzyl-α-d-xylofuranose (α,β)-1 (89:11) | ||
| Empirical formula, (weight) | C26H28O5, (420.48) | ||
| Temperature (K) | 100(2) | ||
| Diffractometer Rigaku® | XtaLabPro mm003+Pilatus200k | ||
| Wavelength (Å) | 0.71073 | ||
| Crystal system, space group | P212121, Orthorhombic | ||
| Unit cell dimensions (Å, α = β = γ = 90°) | a = 6.9002(1) b = 13.1783(2) c = 23.9751(5) | ||
| Volume (Å3) | 2180.13(6) | ||
| Z, Calculated density (Mg/m3) | 4, 1.281 | ||
| Absorption coefficient (mm−1) | 0.088 | ||
| F(000) | 896 | ||
| Theta range for data collection (°) | 2.98 to 30.25 | ||
| Limiting indices | −9 ≤ h ≤ 9, −17 ≤ k ≤ 18, −33 ≤ l ≤ 33 | ||
| Reflections collected/unique R(int) | 55,919/6102 0.0370 | ||
| Completeness to θ max (iUCR)(%) | 99.8 | ||
| Absorption correction method | Gaussian and Multi-scan 1.000 and 0.302 | ||
| Refinement method | IAM Shelxl | HAR NoSpherA2 | |
| Data/restraints/parameters | 6102/416/419 | 6102/484/693 | |
| Goodness-of-fit on F2 | 1.028 | 1.1284 | |
| Final R indices [I > 2σ(I)] | R1, wR2 | 0.0338, 0.0896 | 0.0171, 0.0305 |
| R indices (all data) | R1, wR2 | 0.0352, 0.0903 | 0.0185, 0.0307 |
| Absolute structure parameter [39] | −0.02(15) | 0.07(14) | |
| Largest diff. peak and hole (e.Å−3) | 0.273 and −0.192 | 0.145 and −0.124 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taffoureau, B.; Gillaizeau, I.; Retailleau, P.; Nicolas, C. 2,3,5-Tri-O-benzyl-d-xylofuranose. Molbank 2022, 2022, M1382. https://doi.org/10.3390/M1382
Taffoureau B, Gillaizeau I, Retailleau P, Nicolas C. 2,3,5-Tri-O-benzyl-d-xylofuranose. Molbank. 2022; 2022(2):M1382. https://doi.org/10.3390/M1382
Chicago/Turabian StyleTaffoureau, Baptiste, Isabelle Gillaizeau, Pascal Retailleau, and Cyril Nicolas. 2022. "2,3,5-Tri-O-benzyl-d-xylofuranose" Molbank 2022, no. 2: M1382. https://doi.org/10.3390/M1382
APA StyleTaffoureau, B., Gillaizeau, I., Retailleau, P., & Nicolas, C. (2022). 2,3,5-Tri-O-benzyl-d-xylofuranose. Molbank, 2022(2), M1382. https://doi.org/10.3390/M1382