
6,6′-Di-(2″-thiophenol)-2,2′-bipyridine


{kind=link}
{kind=link}
{kind=link}
Abstract
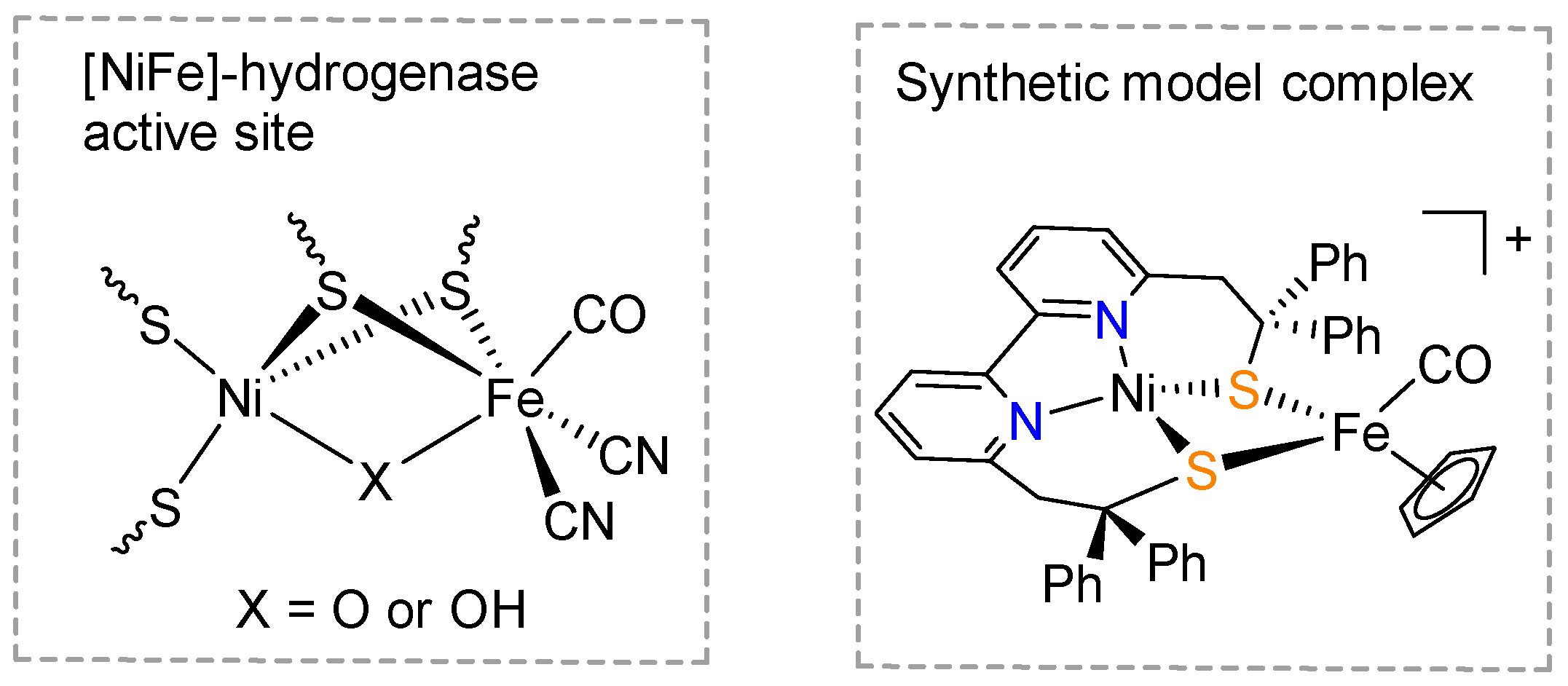
:1. Introduction
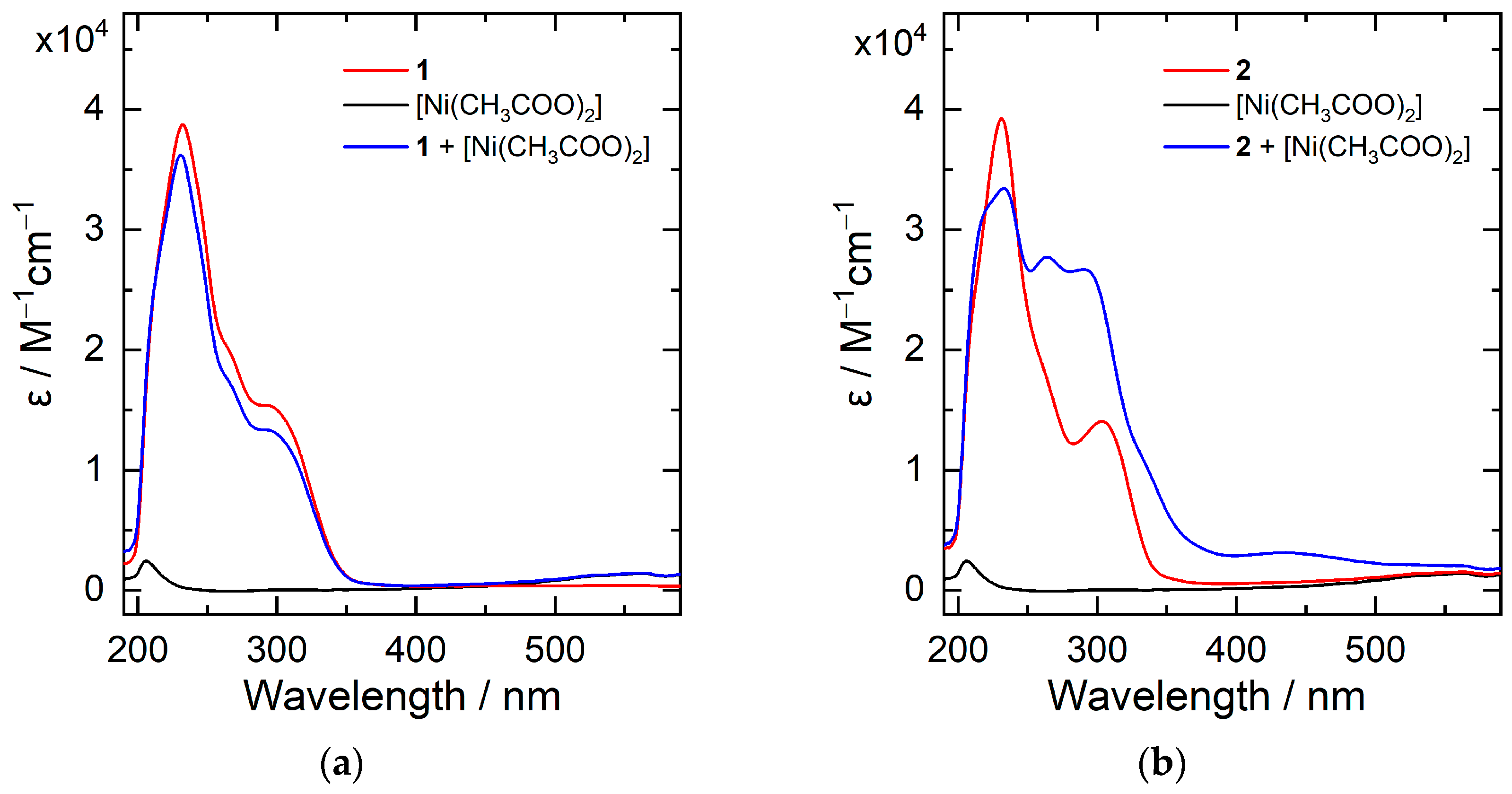
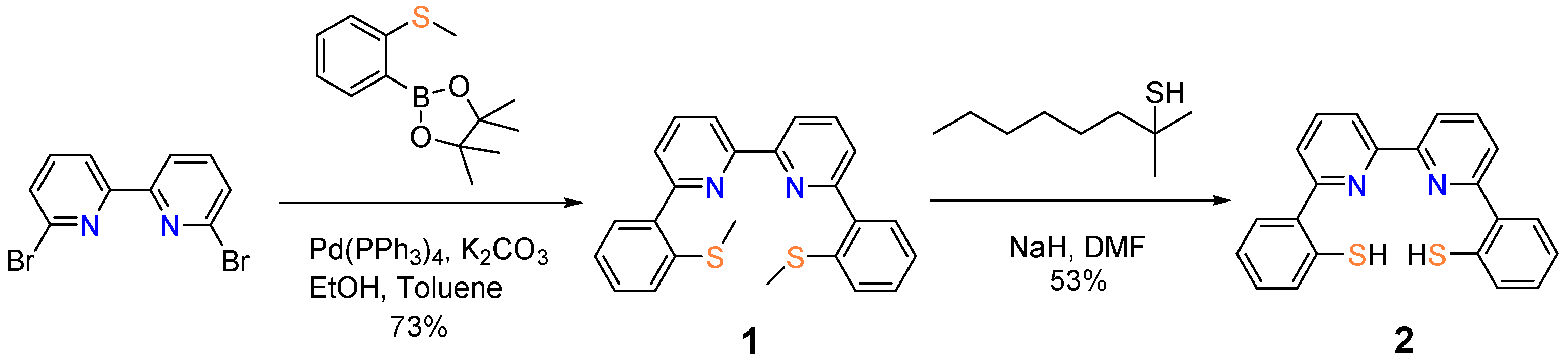
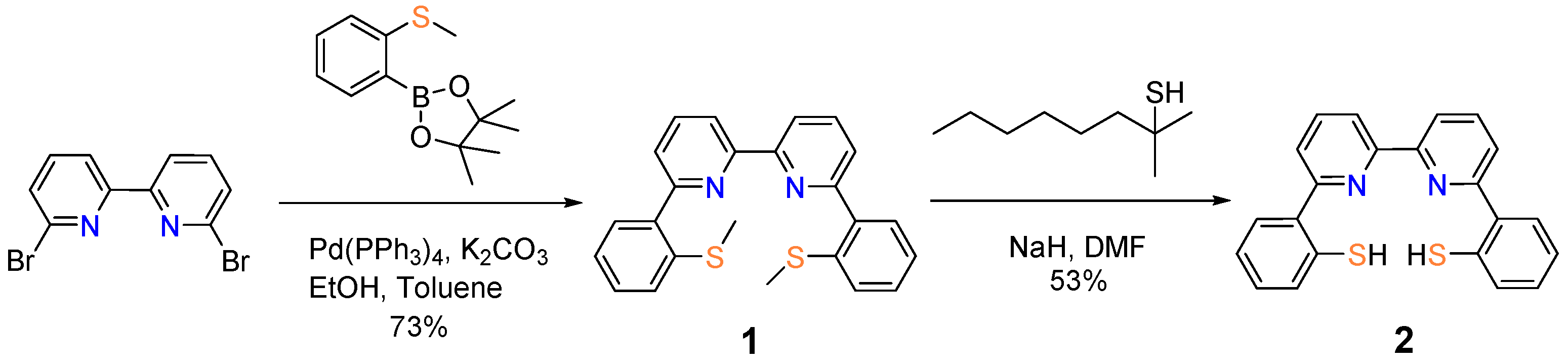
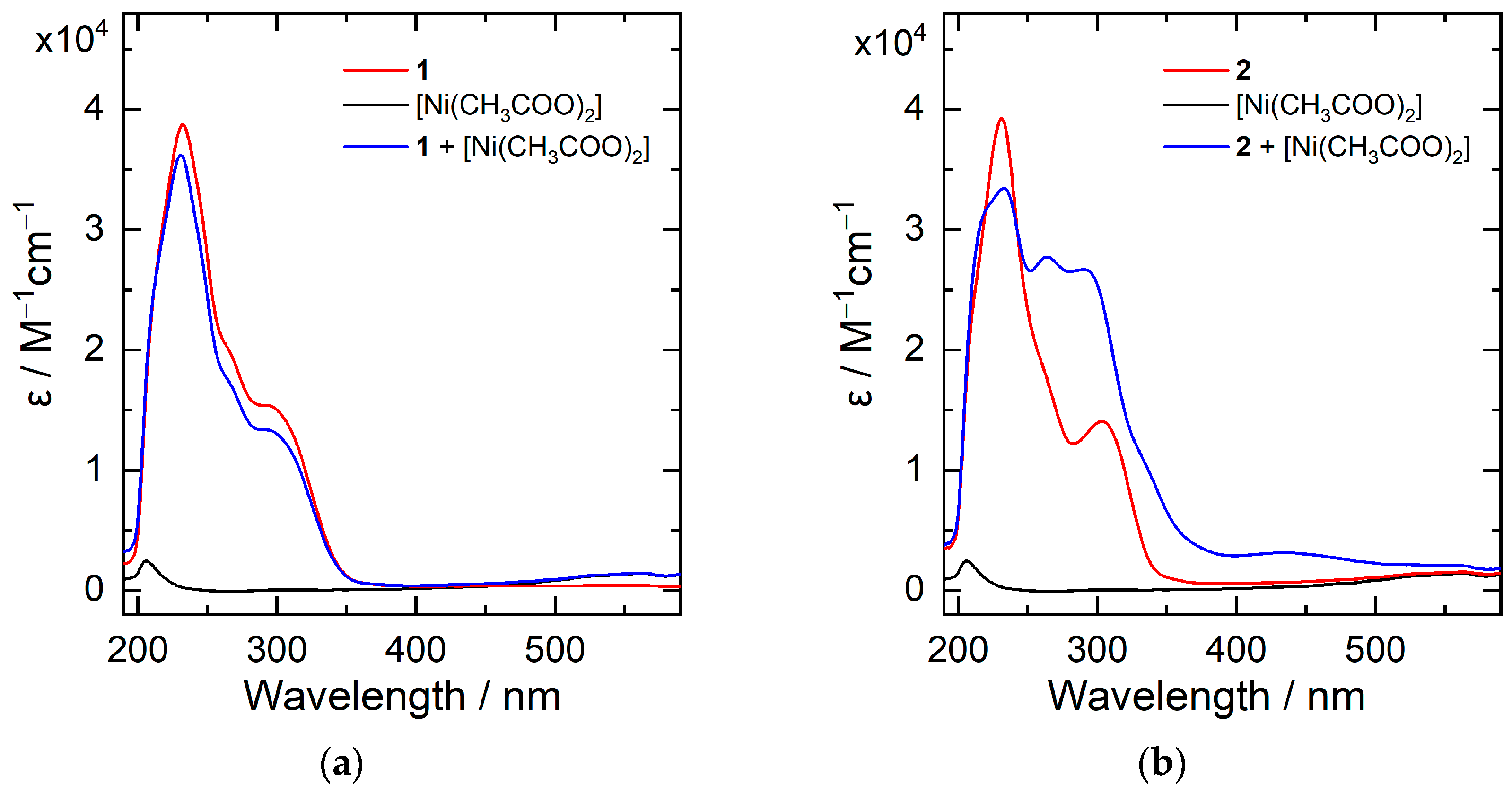
2. Results and Discussion
3. Materials and Methods
- Synthesis of 6,6′-di-(2″-methylthiophenyl)-2,2′-bipyridine (1).
- Synthesis of 6,6′-di-(2″-thiophenol)-2,2′-bipyridine (2).
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Jones, A.K.; Sillery, E.; Albracht, S.P.; Armstrong, F.A. Direct comparison of the electrocatalytic oxidation of hydrogen by an enzyme and a platinum catalyst. Chem. Commun. 2002, 866–867. [Google Scholar] [CrossRef] [PubMed]
- Frey, M. Hydrogenases: Hydrogen-Activating Enzymes. ChemBioChem 2002, 3, 153–160. [Google Scholar] [CrossRef]
- Volbeda, A.; Charon, M.-H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, J.; Scheerer, P.; Frielingsdorf, S.; Kroschinsky, S.; Friedrich, B.; Lenz, O.; Spahn, C.M.T. The crystal structure of an oxygen-tolerant hydrogenase uncovers a novel iron-sulphur centre. Nature 2011, 479, 249. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Nishikawa, K.; Lubitz, W. Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase. Nature 2015, 520, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Simmons, T.R.; Berggren, G.; Bacchi, M.; Fontecave, M.; Artero, V. Mimicking hydrogenases: From biomimetics to artificial enzymes. Coord. Chem. Rev. 2014, 270–271, 127–150. [Google Scholar] [CrossRef]
- Ahmed, M.E.; Dey, A. Recent developments in bioinspired modelling of [NiFe]- and [FeFe]-hydrogenases. Curr. Opin. Electrochem. 2019, 15, 155–164. [Google Scholar] [CrossRef]
- Staffell, I.; Scamman, D.; Abad, A.V.; Balcombe, P.; Dodds, P.E.; Ekins, P.; Shah, N.; Ward, K.R. The role of hydrogen and fuel cells in the global energy system. Energy Environ. Sci. 2019, 12, 463–491. [Google Scholar] [CrossRef] [Green Version]
- Brazzolotto, D.; Gennari, M.; Queyriaux, N.; Simmons, T.R.; Pecaut, J.; Demeshko, S.; Meyer, F.; Orio, M.; Artero, V.; Duboc, C. Nickel-centred proton reduction catalysis in a model of [NiFe] hydrogenase. Nat. Chem. 2016, 8, 1054–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.E.; Chattopadhyay, S.; Wang, L.; Brazzolotto, D.; Pramanik, D.; Aldakov, D.; Fize, J.; Morozan, A.; Gennari, M.; Duboc, C.; et al. Hydrogen Evolution from Aqueous Solutions Mediated by a Heterogenized [NiFe]-Hydrogenase Model: Low pH Enables Catalysis through an Enzyme-Relevant Mechanism. Angew. Chem. Int. Ed. 2018, 57, 16001–16004. [Google Scholar] [CrossRef] [PubMed]
- Brazzolotto, D.; Wang, L.; Tang, H.; Gennari, M.; Queyriaux, N.; Philouze, C.; Demeshko, S.; Meyer, F.; Orio, M.; Artero, V.; et al. Tuning Reactivity of Bioinspired [NiFe]-Hydrogenase Models by Ligand Design and Modeling the CO Inhibition Process. ACS Catal. 2018, 8, 10658–10667. [Google Scholar] [CrossRef]
- Gennari, M.; Orio, M.; Pecaut, J.; Neese, F.; Collomb, M.N.; Duboc, C. Reversible apical coordination of imidazole between the Ni(III) and Ni(II) oxidation states of a dithiolate complex: A process related to the Ni superoxide dismutase. Inorg. Chem. 2010, 49, 6399–6401. [Google Scholar] [CrossRef] [PubMed]
- Hamacher, C.; Hurkes, N.; Kaiser, A.; Klein, A.; Schuren, A. Electrochemistry and spectroscopy of organometallic terpyridine nickel complexes. Inorg. Chem. 2009, 48, 9947–9951. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K. White Organic Electroluminescence Device. JP Patent JP2010135689A, 17 June 2010. [Google Scholar]
- Neuhaus, J.D.; Morrow, S.M.; Brunavs, M.; Willis, M.C. Diversely Substituted Quinolines via Rhodium-Catalyzed Alkyne Hydroacylation. Org. Lett. 2016, 18, 1562–1565. [Google Scholar] [CrossRef] [Green Version]
- Manes, T.A.; Rose, M.J. Mono- and Dinuclear Manganese Carbonyls Supported by 1,8-Disubstituted (L = Py, SMe, SH) Anthracene Ligand Scaffolds. Inorg. Chem. 2016, 55, 5127–5138. [Google Scholar] [CrossRef]
- Kim, H.J.; Yoon, J.H.; Yoon, S. Photooxidative coupling of thiophenol derivatives to disulfides. J. Phys. Chem. A 2010, 114, 12010–12015. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Tong, L. 6,6′-Di-(2″-thiophenol)-2,2′-bipyridine. Molbank 2022, 2022, M1355. https://doi.org/10.3390/M1355
Huang Y, Tong L. 6,6′-Di-(2″-thiophenol)-2,2′-bipyridine. Molbank. 2022; 2022(2):M1355. https://doi.org/10.3390/M1355
Chicago/Turabian StyleHuang, Yan, and Lianpeng Tong. 2022. "6,6′-Di-(2″-thiophenol)-2,2′-bipyridine" Molbank 2022, no. 2: M1355. https://doi.org/10.3390/M1355
APA StyleHuang, Y., & Tong, L. (2022). 6,6′-Di-(2″-thiophenol)-2,2′-bipyridine. Molbank, 2022(2), M1355. https://doi.org/10.3390/M1355