
New Derivatives of Lupeol and Their Biological Activity

{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
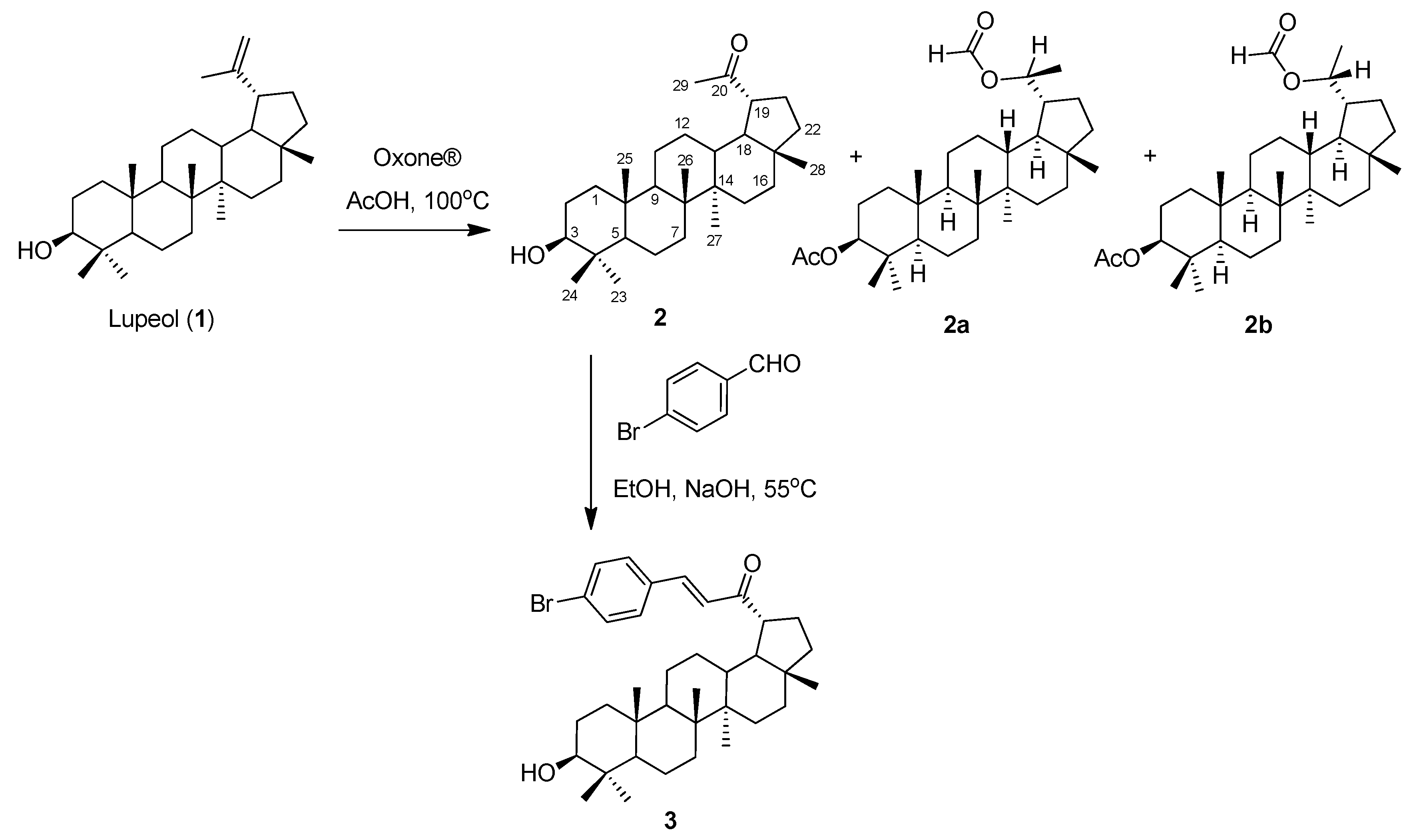
2.1. Synthesis
2.2. α-Glucosidase Inhibition of 2a, 2b, and 3
3. Materials and Methods
3.1. Materials
3.2. Synthesis Procedure
3.3. α-Glucosidase Inhibitory Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tabish, S.A. Is diabetes becoming the biggest epidemic of the twenty-first century? Int. J. Health Sci. 2007, 1, 3–8. [Google Scholar]
- Gallo, M.B.C.; Sarachine, M.J. Biological activities of lupeol. Int. J. Biomed. Pharm. Sci. 2009, 3, 46–66. [Google Scholar]
- Starks, C.M.; Williams, R.B.; Norman, V.L.; Lawrence, J.A.; Goering, M.G.; O’Neil-Johnson, M.; Hu, J.F.; Rice, S.M.; Eldridge, G.R. Abronione, a rotenoid from the desert annual Abronia villosa. Phytochem. Lett. 2011, 4, 72–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddique, H.R.; Saleem, M. Beneficial health effects of lupeol triterpene: A review of preclinical studies. Life Sci. 2011, 88, 285–293. [Google Scholar] [CrossRef]
- Papi Reddy, K.; Singh, A.B.; Puri, A.; Srivastava, A.K.; Narender, T. Synthesis of novel triterpenoid (lupeol) derivatives and their in vivo antihyperglycemic and antidyslipidemic activity. Bioorg. Med. Chem. Lett. 2009, 19, 4463–4466. [Google Scholar] [CrossRef]
- Phan, H.V.T.; Duong, T.H.; Pham, D.D.; Pham, H.A.; Nguyen, V.K.; Nguyen, T.P.; Nguyen, H.H.; Nguyen, N.H.; Sam-ang, P.; Phontree, K.; et al. Design and synthesis of new lupeol derivatives and their α-glucosidase inhibitory and cytotoxic activities. Nat. Prod. Res. 2020, Article in press. [Google Scholar] [CrossRef] [PubMed]
- Sichaem, J.; Aree, T.; Lugsanangarm, K.; Tip-Pyang, S. Identification of highly potent α-glucosidase inhibitory and antioxidant constituents from Zizyphus rugosa bark: Enzyme kinetic and molecular docking studies with active metabolites. Pharm. Biol. 2017, 55, 1436–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sichaem, J.; Inthanon, K.; Funnimid, N.; Phontree, K.; Phan, H.V.T.; Tran, T.M.D.; Niamnont, N.; Srikittiwanna, K.; Sedlak, S.; Duong, T.H. Chemical constituents of the stem bark of Bombax ceiba. Chem. Nat. Compd. 2020, 56, 909–911. [Google Scholar] [CrossRef]
- Uyanik, M.; Akakura, M.; Ishihara, K. 2-Iodoxybenzenesulfonic acid as an extremely active catalyst for the selective oxidation of alcohols to aldehydes, ketones, carboxylic acids, and enones with Oxone. J. Am. Chem. Soc. 2009, 131, 251–262. [Google Scholar] [CrossRef]
- Corbett, R.E.; Cong, A.N.T.; Wilkins, A.L.; Thomson, R.A. Lichens and Fungi. Part 17. The synthesis and absolute configuration at C-20 of the (R)- and (S)-epimers of some 29-substituted lupane derivatives and of some 30-norlupan-20-ol derivatives and the crystal structure of (20R)-3β-acetoxylupan-29-ol. J. Chem. Soc. Perkin Trans. 1985, 17, 2051–2056. [Google Scholar] [CrossRef]
- Corbett, R.E.; Cong, A.N.T.; Holland, P.T.; Wilkins, A.L. Lichens and Fungi. XVIII. Extractives from Pseudocyphellaria rubella. Aust. J. Chem. 1987, 40, 461–468. [Google Scholar] [CrossRef]
- Dao, T.B.N.; Nguyen, T.M.T.; Nguyen, V.Q.; Tran, T.M.D.; Tran, N.M.A.; Nguyen, C.H.; Nguyen, T.H.T.; Nguyen, H.H.; Sichaem, J.; Tran, C.L.; et al. Flavones from Combretum quadrangulare growing in Vietnam and their alpha-glucosidase inhibitory activity. Molecules 2021, 26, 2531. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Le, H.-T.-T.; Chau, Q.-C.; Duong, T.-H.; Tran, Q.-T.-P.; Pham, N.-K.-T.; Nguyen, T.-H.-T.; Nguyen, N.-H.; Sichaem, J. New Derivatives of Lupeol and Their Biological Activity. Molbank 2021, 2021, M1306. https://doi.org/10.3390/M1306
Le H-T-T, Chau Q-C, Duong T-H, Tran Q-T-P, Pham N-K-T, Nguyen T-H-T, Nguyen N-H, Sichaem J. New Derivatives of Lupeol and Their Biological Activity. Molbank. 2021; 2021(4):M1306. https://doi.org/10.3390/M1306
Chicago/Turabian StyleLe, Hoang-Thuy-Tien, Quoc-Cuong Chau, Thuc-Huy Duong, Quyen-Thien-Phuc Tran, Nguyen-Kim-Tuyen Pham, Thi-Hoai-Thu Nguyen, Ngoc-Hong Nguyen, and Jirapast Sichaem. 2021. "New Derivatives of Lupeol and Their Biological Activity" Molbank 2021, no. 4: M1306. https://doi.org/10.3390/M1306
APA StyleLe, H.-T.-T., Chau, Q.-C., Duong, T.-H., Tran, Q.-T.-P., Pham, N.-K.-T., Nguyen, T.-H.-T., Nguyen, N.-H., & Sichaem, J. (2021). New Derivatives of Lupeol and Their Biological Activity. Molbank, 2021(4), M1306. https://doi.org/10.3390/M1306