
Practical and Asymmetric Synthesis of Apremilast Using Ellman’s Sulfinamide as a Chiral Auxiliary


Abstract
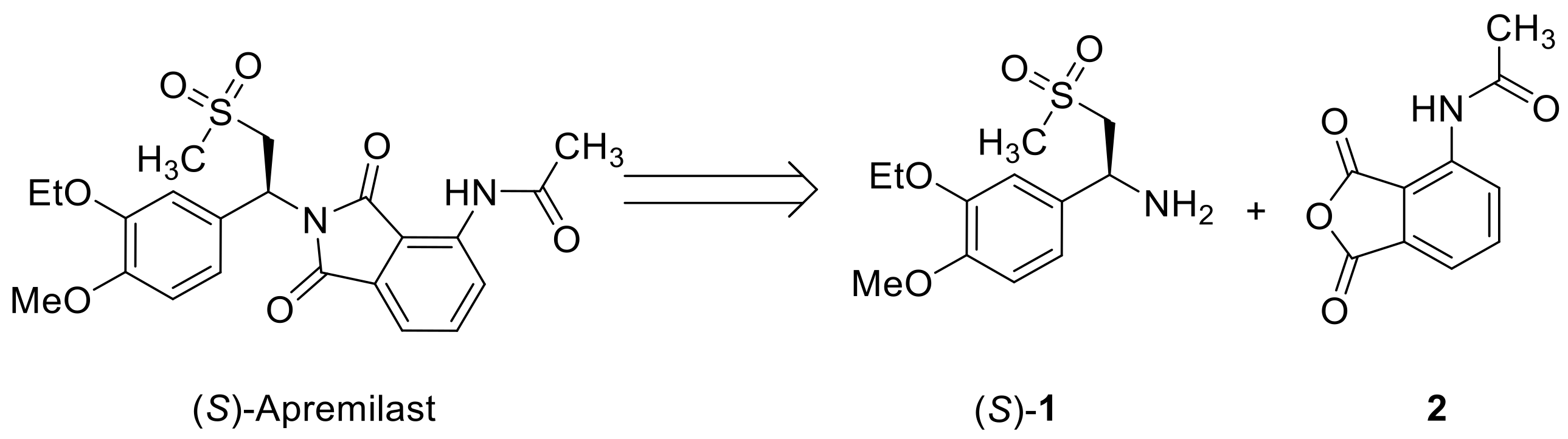
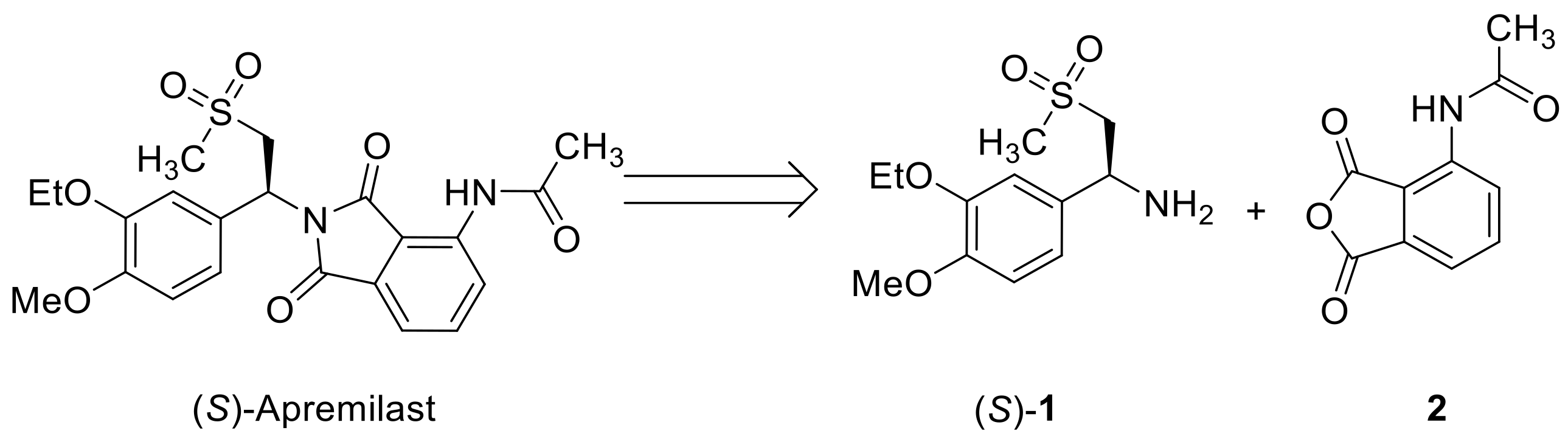
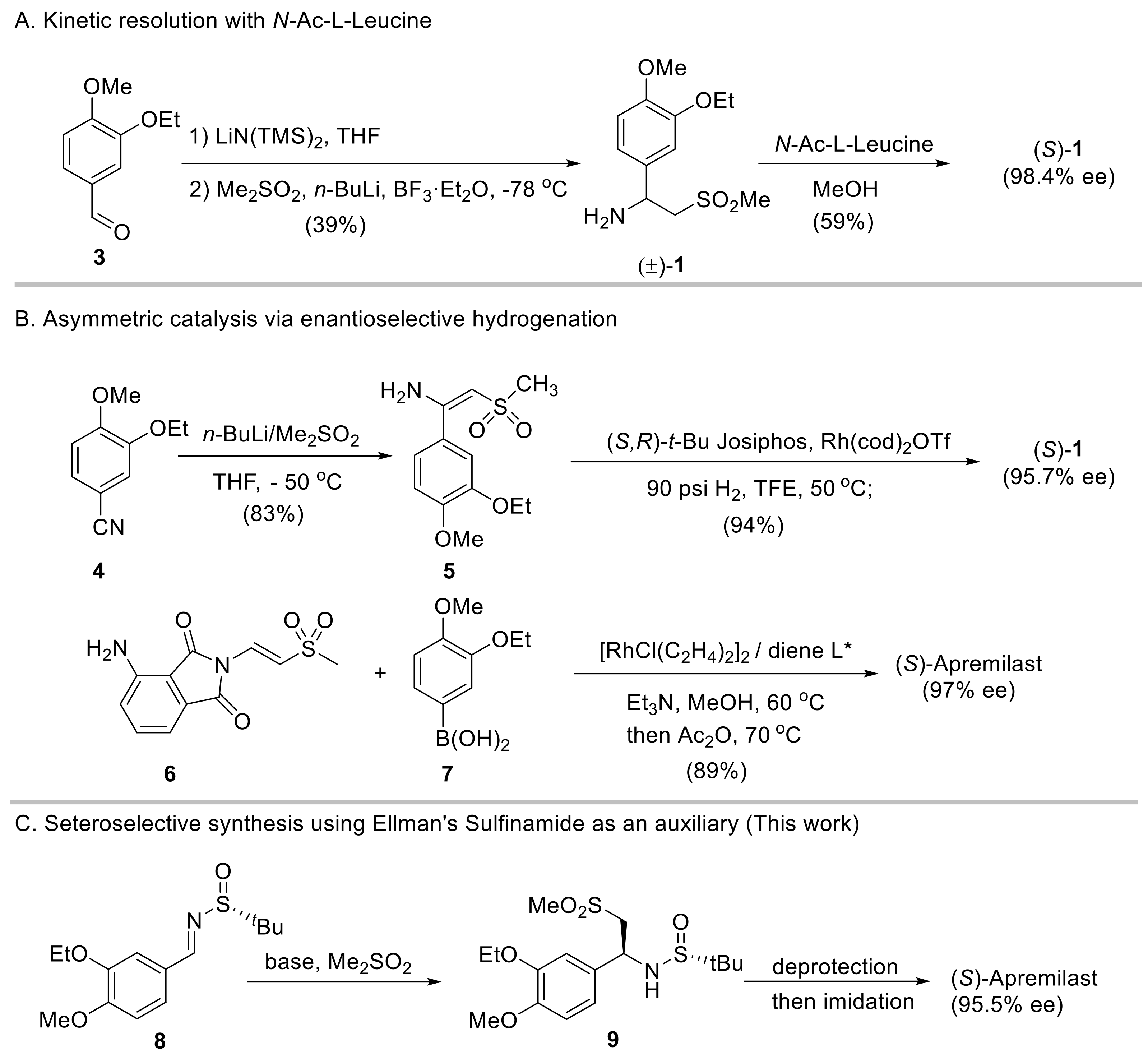
:1. Introduction
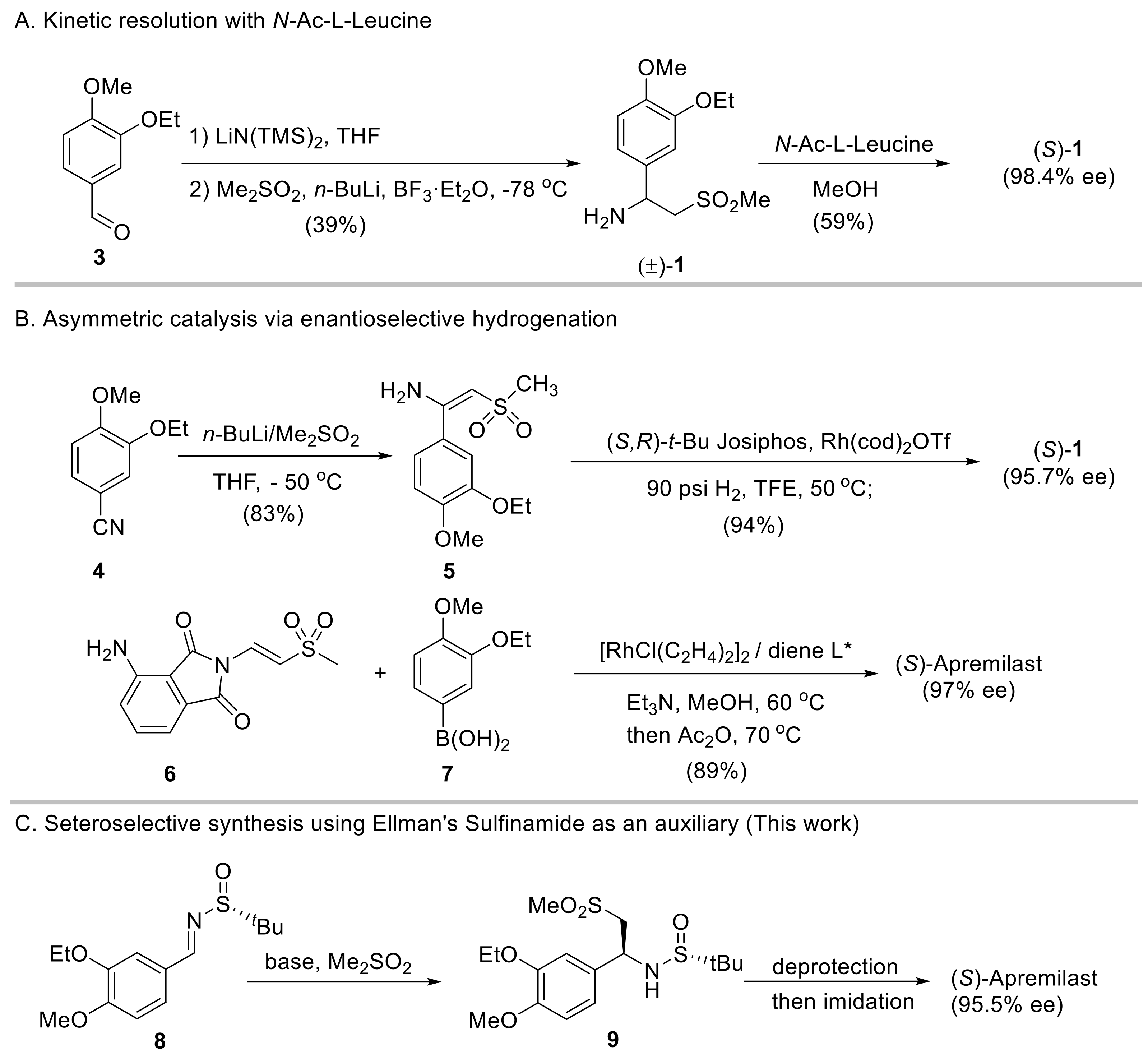
2. Results and Discussion
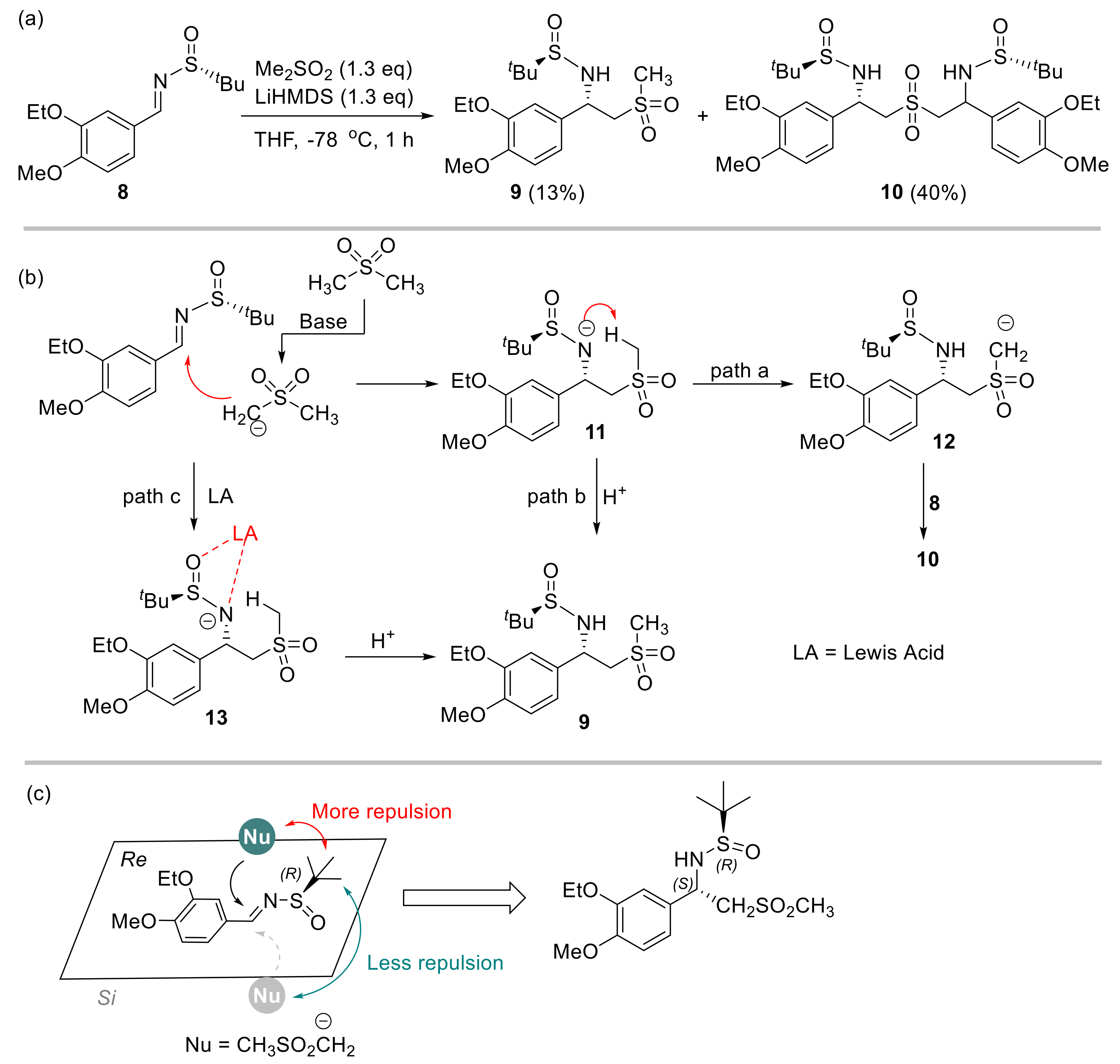
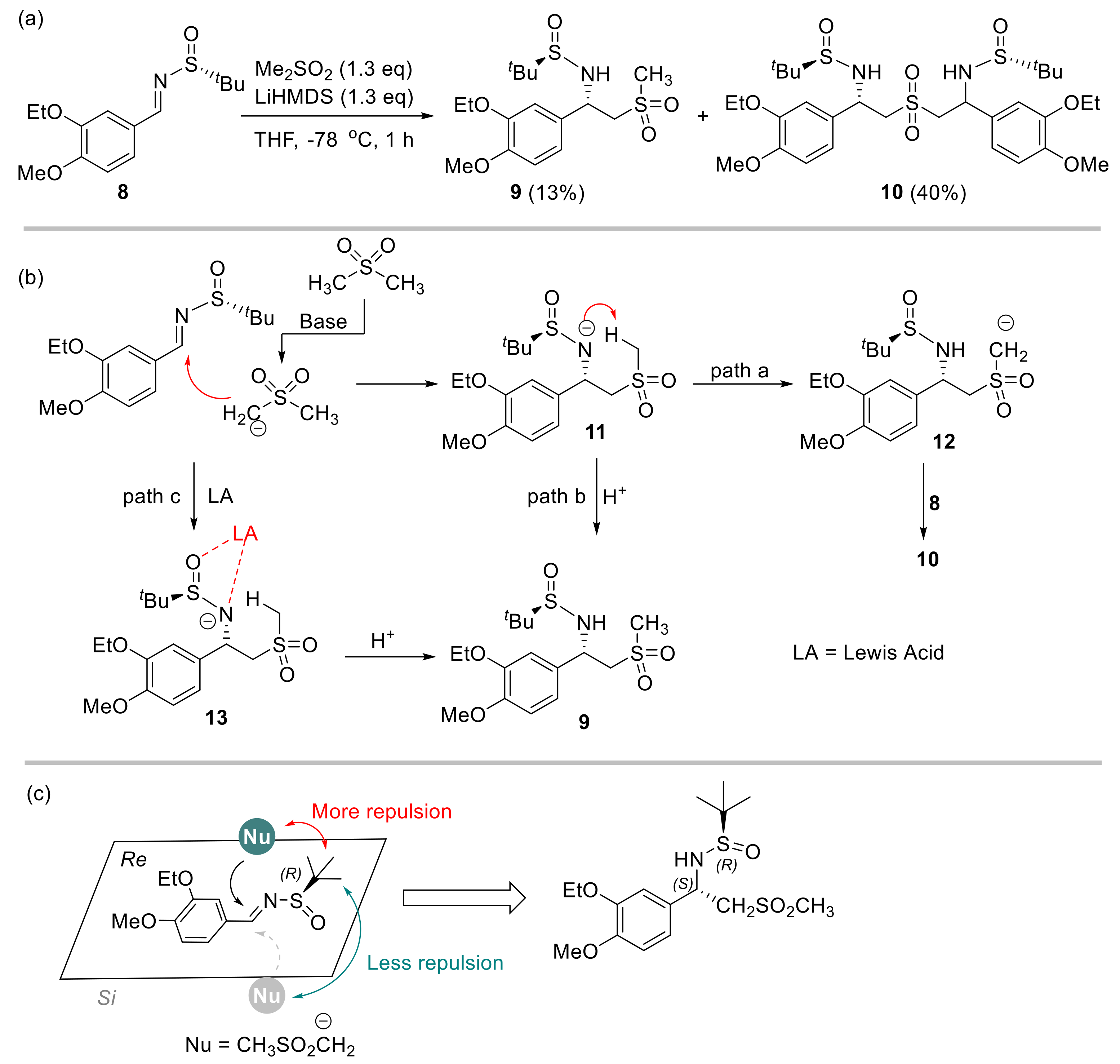
2.1. Initial Reaction Attempt and Rational of Outcome
2.2. Reaction Condition Optmizations
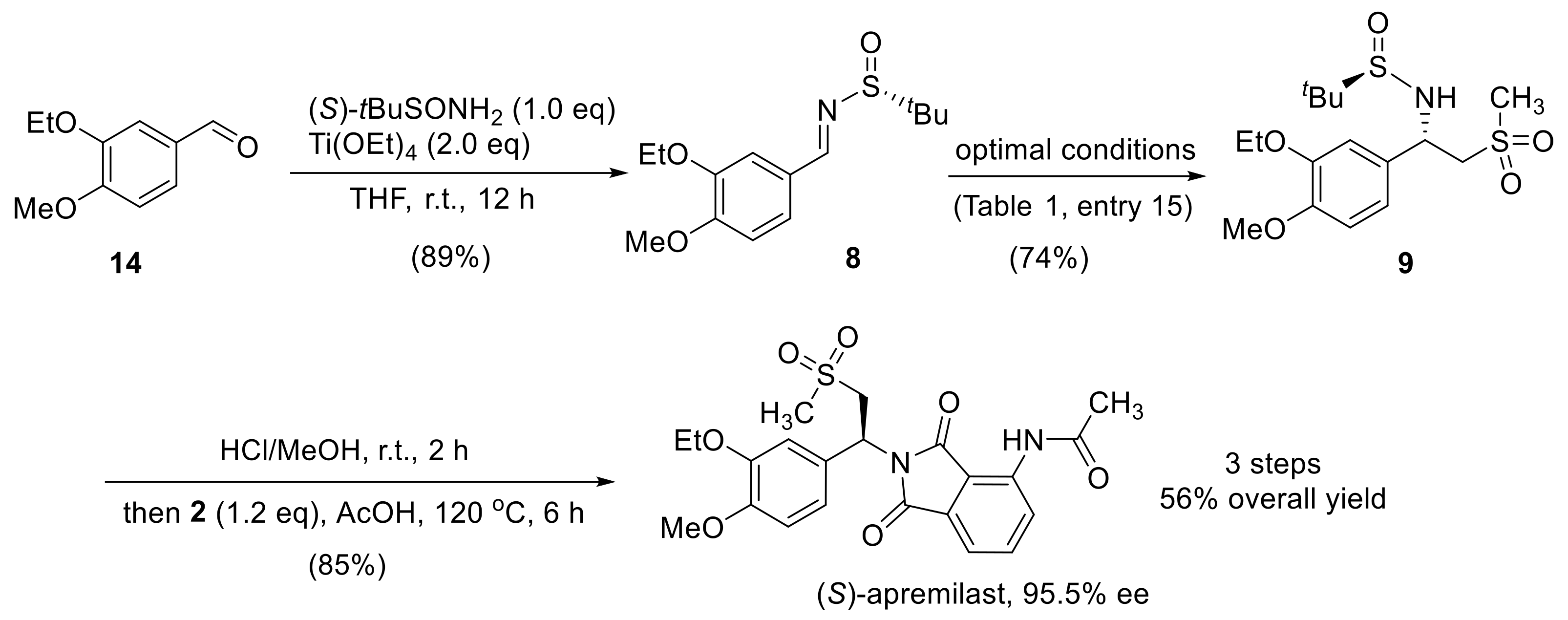
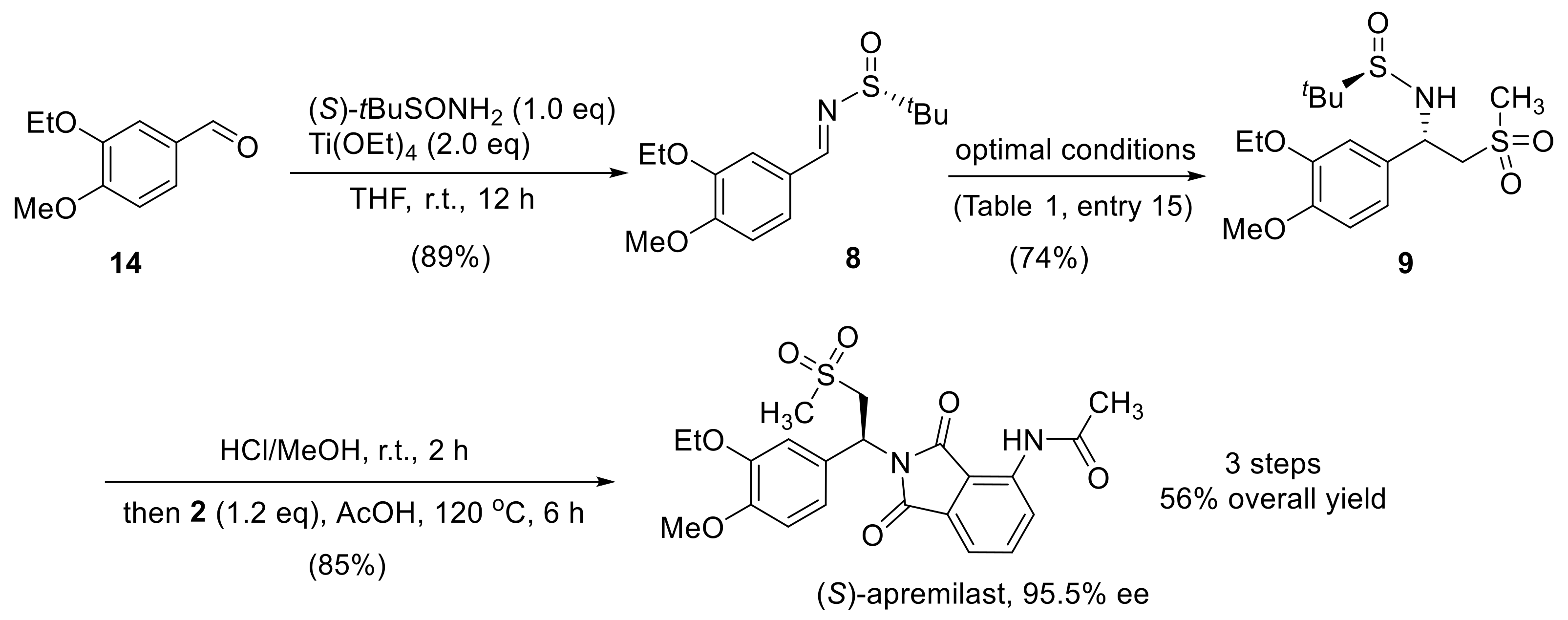
2.3. Synthesis of Apremilast
3. Materials and Methods
3.1. General
3.2. Synthesis of (R,E)-N-(3-Ethoxy-4-Methoxybenzylidene)-2-Methylpropane-2-Sulfinamide (8)
3.3. Synthesis of (R)-N-((S)-1-(3-Ethoxy-4-Methoxyphenyl)-2-(Methylsulfonyl)ethyl)-2-Methylpropane-2-Sulfinamide (9)
3.4. Synthesis of (S)-N-(2-(1-(3-Ethoxy-4-Methoxyphenyl) -2-(Methylsulfonyl)ethyl)-1,3-Dioxoisoindolin-4-yl)Acetamide (Apremilast)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Papp, K.; Cather, J.C.; Rosoph, L.; Sofen, H.; Langley, R.G.; Matheson, R.T.; Hu, C.C.; Day, R.M. Efficacy of apremilast in the treatment of moderate to severe psoriasis: A randomised controlled trial. Lancet 2012, 380, 738–746. [Google Scholar] [CrossRef]
- US Food and Drug Administration. FDA News Release: FDA Approves Otezla to Treat Psoriatic Arthritis. US Department of Health and Human Services. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=205437 (accessed on 25 August 2021).
- Pages, L.; Gavaldà, A.; Lehner, M.D. PDE4 inhibitors: A review of current developments (2005–2009). Expert Opin. Ther. Pat. 2009, 19, 1501–1519. [Google Scholar] [CrossRef] [PubMed]
- Schafer, P. Apremilast mechanism of action and application to psoriasis and psoriatic arthritis. Biochem. Pharmacol. 2012, 83, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Meadows, D.C.; Gervay-Hague, J. Vinyl sulfones: Synthetic preparations and medicinal chemistry applications. Med. Res. Rev. 2006, 26, 793–814. [Google Scholar] [CrossRef] [PubMed]
- Back, T.G. Design and synthesis of some biologically interesting natural and unnatural products based on organosulfur and selenium chemistry. Can. J. Chem. 2009, 87, 1657–1674. [Google Scholar] [CrossRef]
- Ghislieri, D.; Turner, N.J. Biocatalytic approaches to the synthesis of enantiomerically pure chiral amines. Top. Catal. 2014, 57, 284–300. [Google Scholar] [CrossRef]
- Guo, F.; Berglund, P. Transaminase biocatalysis: Optimization and application. Green Chem. 2017, 19, 333–360. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Shi, Y.; Zhong, F. Rhodium-catalyzed intermolecular C(sp3)–H amination in a purely aqueous system. Green. Chem. 2018, 20, 113–117. [Google Scholar] [CrossRef]
- Michael, F.; Farnberger, J.E.; Kroutil, W. The Industrial Age of Biocatalytic Transamination. Eur. J. Org. Chem. 2015, 32, 6965–6982. [Google Scholar]
- Kelly, S.A.; Pohle, S.; Wharry, S.; Mix, S.; Allen, C.C.R.; Moody, T.S.; Gilmore, B.F. Application of ω-Transaminases in the Pharmaceutical Industry. Chem. Rev. 2018, 118, 349–367. [Google Scholar] [CrossRef] [PubMed]
- Narode, H.; Gayke, M.; Eppa, G.; Yadav, J.S. A Review on Synthetic Advances toward the Synthesis of Apremilast, an Anti-inflammatory Drug. Org. Process. Res. Dev. 2021, 25, 1512–1523. [Google Scholar] [CrossRef]
- Man, H.-W.; Schafer, P.; Wong, L.M.; Patterson, R.T.; Corral, L.G.; Raymon, H.; Blease, K.; Leisten, J.; Shirley, M.A.; Tang, Y.; et al. Dis-covery of (S)-N-{2-[1-(3-Ethoxy-4-methoxyphenyl)-2-methanesulfonylethyl]-1,3-dioxo-2,3-dihydro-1H-isoindol-4-yl}acetamide (Apremilast), a Potent and Orally Active Phosphodiesterase 4 and Tumor Necrosis Factor-α Inhibitor. J. Med. Chem. 2009, 52, 1522–1524. [Google Scholar] [CrossRef] [PubMed]
- Ruchelman, A.L.; Connolly, T.J. Enantioselective synthesis of the apremilast aminosulfone using catalytic asymmetric hydrogenation. Tetrahedron Asymmetry 2015, 26, 553–559. [Google Scholar] [CrossRef]
- Gao, W.; Lv, H.; Zhang, X. Rh/DuanPhos-Catalyzed Asymmetric Hydrogenation of β-Acetylamino Vinylsulfides: An Approach to Chiral β-Acetylamino Sulfides. Org. Lett. 2017, 19, 2877–2880. [Google Scholar] [CrossRef] [PubMed]
- Syu, J.-F.; Gopula, B.; Jian, J.-H.; Li, W.-S.; Kuo, T.-S.; Wu, P.-Y.; Henschke, J.P.; Hsieh, M.-C.; Tsai, M.-K.; Wu, H.-L. Asymmetric Synthesis of β-Aryl β-Imido Sulfones Using Rhodium Catalysts with Chiral Diene Ligands: Synthesis of Apremilast. Org. Lett. 2019, 21, 4614–4618. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Cogan, D.A.; Ellman, J.A. Catalytic Asymmetric Synthesis of tert-Butanesulfinamide. Application to the Asymmetric Synthesis of Amines. J. Am. Chem. Soc. 1997, 119, 9913–9914. [Google Scholar] [CrossRef]
- Robak, M.T.; Herbage, M.A.; Ellman, J.A. Synthesis and Applications of tert-Butanesulfinamide. Chem. Rev. 2010, 110, 3600–3740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Xu, W.; Zheng, W.; Zhou, P.; Sun, Z. Practical and stereoselective synthesis of β-amino sulfones from alkyl phenyl sulfones and N-(tert-butylsulfinyl) aldimines. Org. Biomol. Chem. 2011, 9, 6502–6505. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
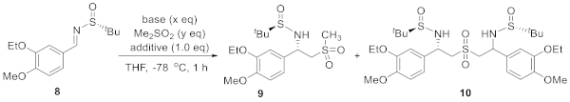
| Entry 1 | Base | x (eq) | y (eq) | Additive | Conv. 2 | 9:10 2 |
|---|---|---|---|---|---|---|
| 1 | LiHMDS | 1.3 | 1.3 | / | >99% | 1:2.5 |
| 2 | NaHMDS | 1.3 | 1.3 | / | >99% | 1:3.2 |
| 3 | n-BuLi | 1.3 | 1.3 | / | >99% | 1:4.7 |
| 4 3 | NaH | 1.3 | 1.3 | / | <10% | / |
| 5 | LiHMDS | 3.0 | 3.0 | / | >99% | 1:1.85 |
| 6 | LiHMDS | 3.0 | 10.0 | / | >99% | 1:0.63 |
| 7 | LiHMDS | 5.0 | 10.0 | / | >99% | 1:0.61 |
| 8 | LiHMDS | 3.0 | 10.0 | MgCl2 | >99% | 1:0.55 |
| 9 | LiHMDS | 3.0 | 10.0 | AlCl3 | >99% | 1:0.3 |
| 10 | LiHMDS | 3.0 | 10.0 | FeCl2 | >99% | 1:3.25 |
| 11 | LiHMDS | 3.0 | 10.0 | NiCl2 | >99% | 1:0.23 |
| 12 | LiHMDS | 3.0 | 10.0 | CuCl2 | >99% | 1:1.15 |
| 13 | LiHMDS | 3.0 | 10.0 | BF3-Et2O | >99% | 1:1.45 |
| 14 | LiHMDS | 3.0 | 10.0 | LiCl | >99% | 1:0.15 |
| 15 4 | LiHMDS | 3.0 | 10.0 | LiCl | >99% | 1:0.15 |
| 16 5 | LiHMDS | 3.0 | 10.0 | LiCl | >99% | 1:0.55 |
| 17 5 | LiHMDS | 1.3 | 10.0 | LiCl | >99% | 1:1.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, B.; Zhong, F. Practical and Asymmetric Synthesis of Apremilast Using Ellman’s Sulfinamide as a Chiral Auxiliary. Molbank 2021, 2021, M1275. https://doi.org/10.3390/M1275
Wang B, Zhong F. Practical and Asymmetric Synthesis of Apremilast Using Ellman’s Sulfinamide as a Chiral Auxiliary. Molbank. 2021; 2021(3):M1275. https://doi.org/10.3390/M1275
Chicago/Turabian StyleWang, Bofei, and Fangrui Zhong. 2021. "Practical and Asymmetric Synthesis of Apremilast Using Ellman’s Sulfinamide as a Chiral Auxiliary" Molbank 2021, no. 3: M1275. https://doi.org/10.3390/M1275
APA StyleWang, B., & Zhong, F. (2021). Practical and Asymmetric Synthesis of Apremilast Using Ellman’s Sulfinamide as a Chiral Auxiliary. Molbank, 2021(3), M1275. https://doi.org/10.3390/M1275