
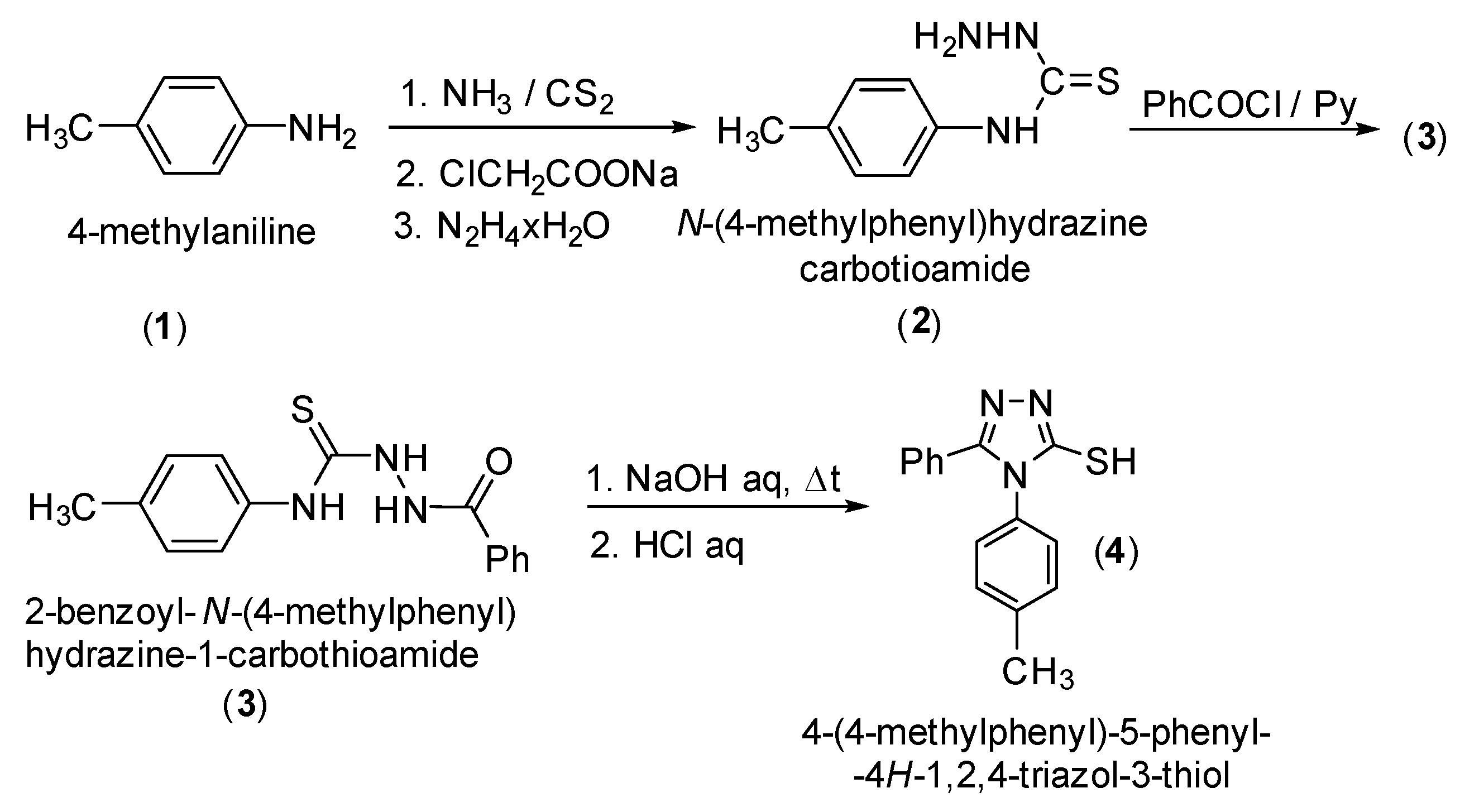
(R,S)-2-{[4-(4-Methylphenyl)-5-phenyl-4H-1,2,4-triazol-3-yl]thio}-1-phenyl-1-ethanol


Abstract
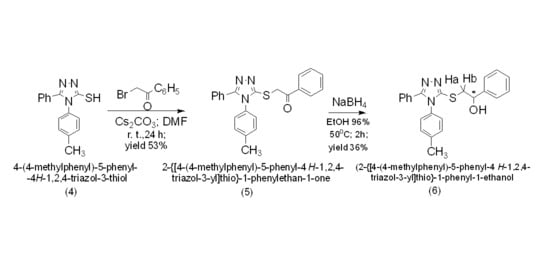
:
1. Introduction
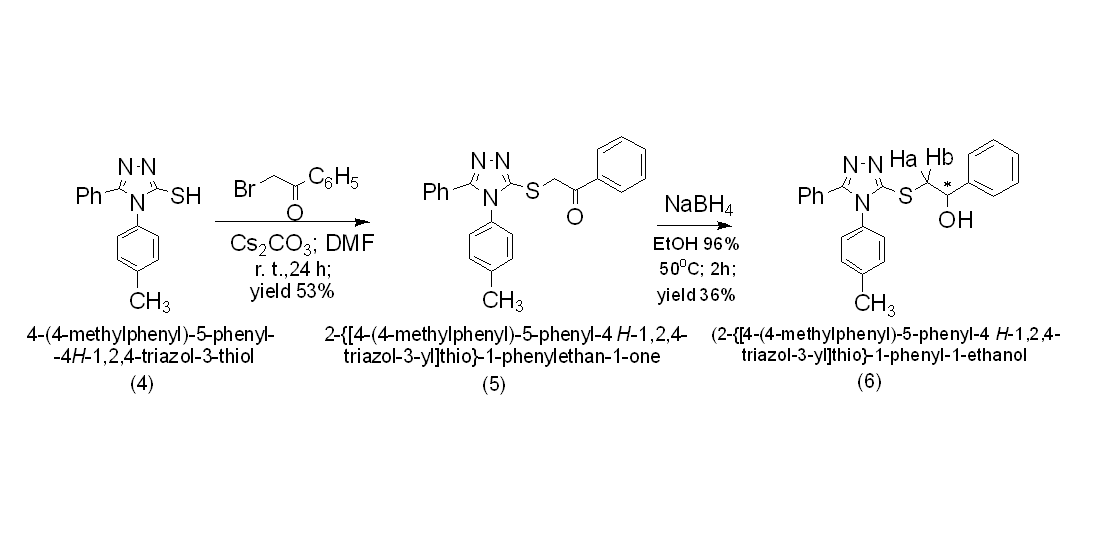
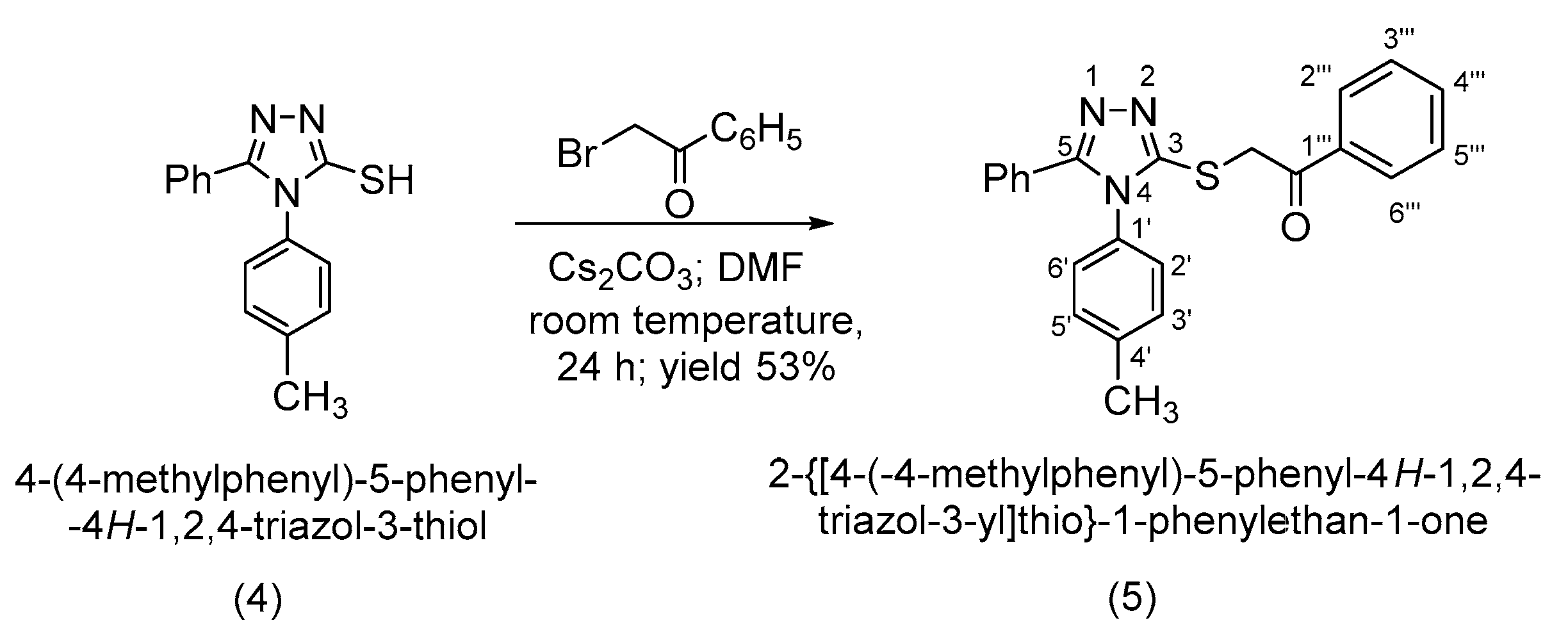
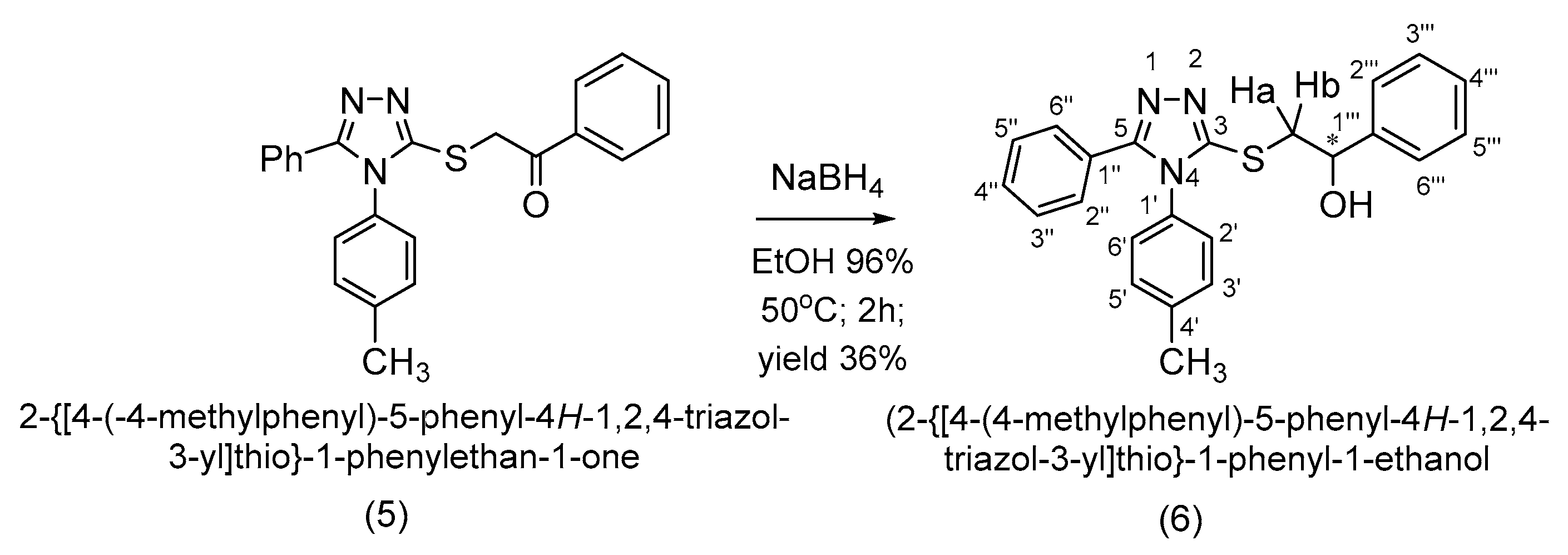
2. Results and Discussion
3. Materials and Methods
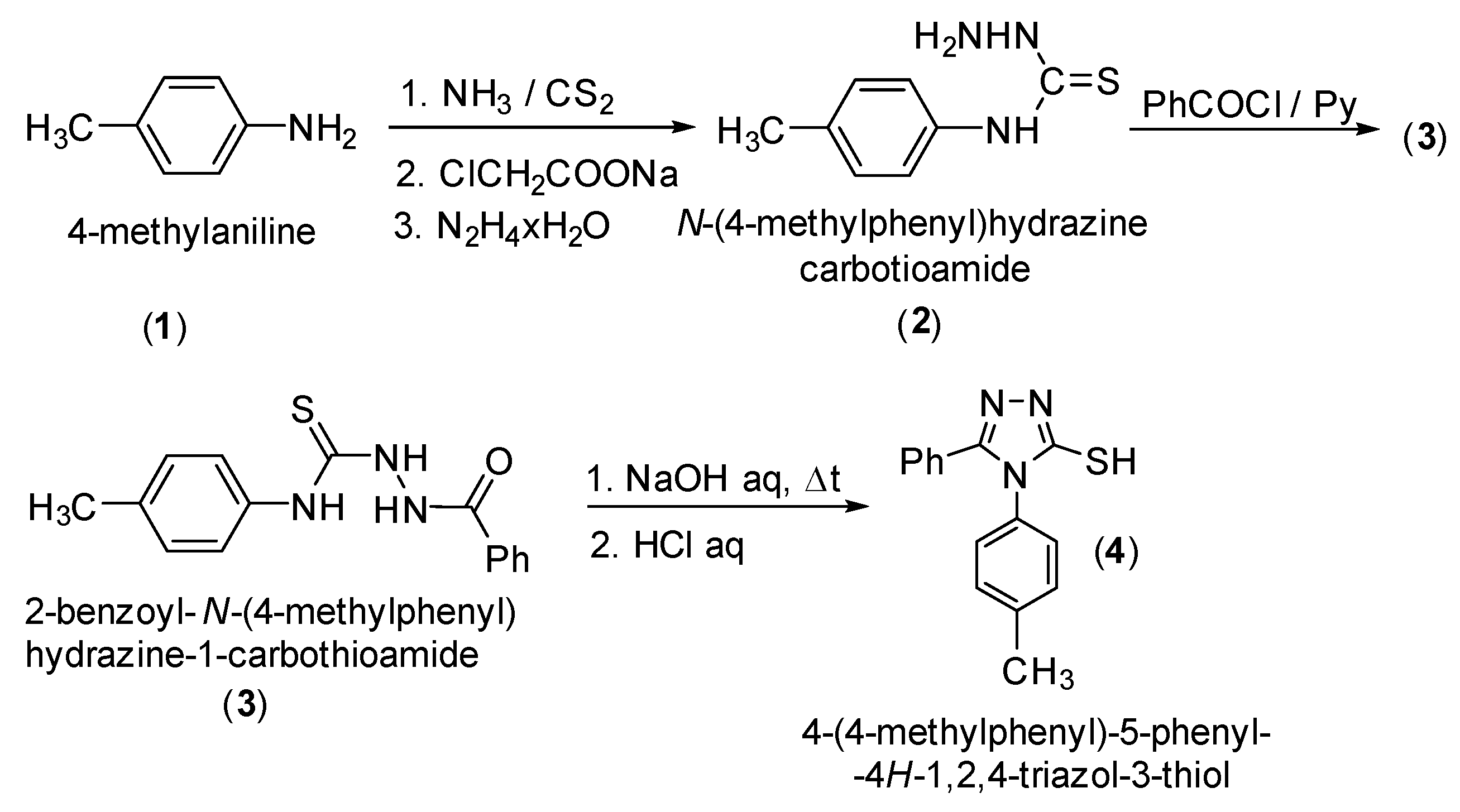
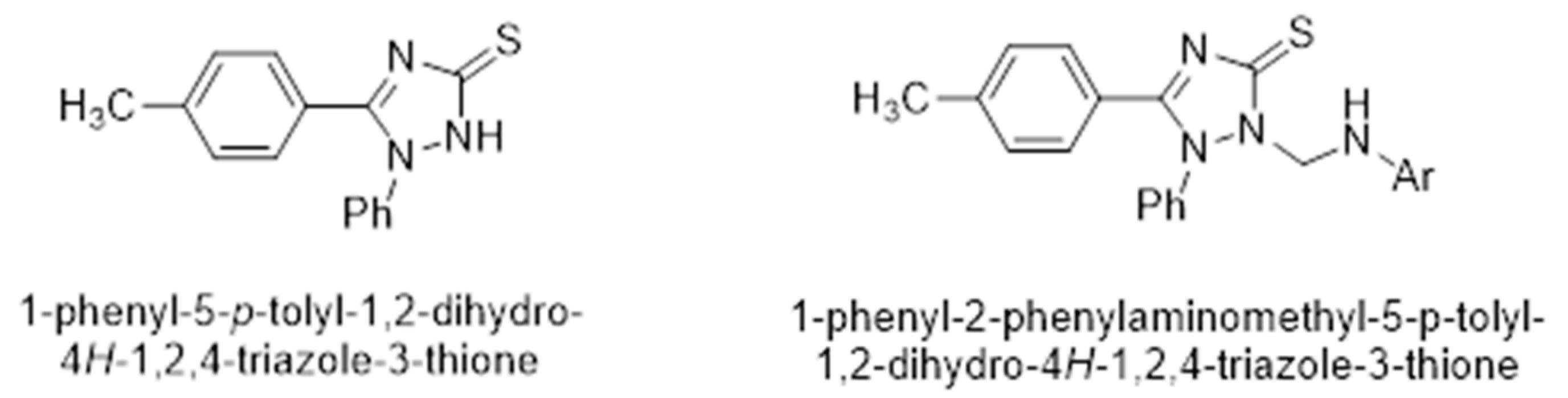
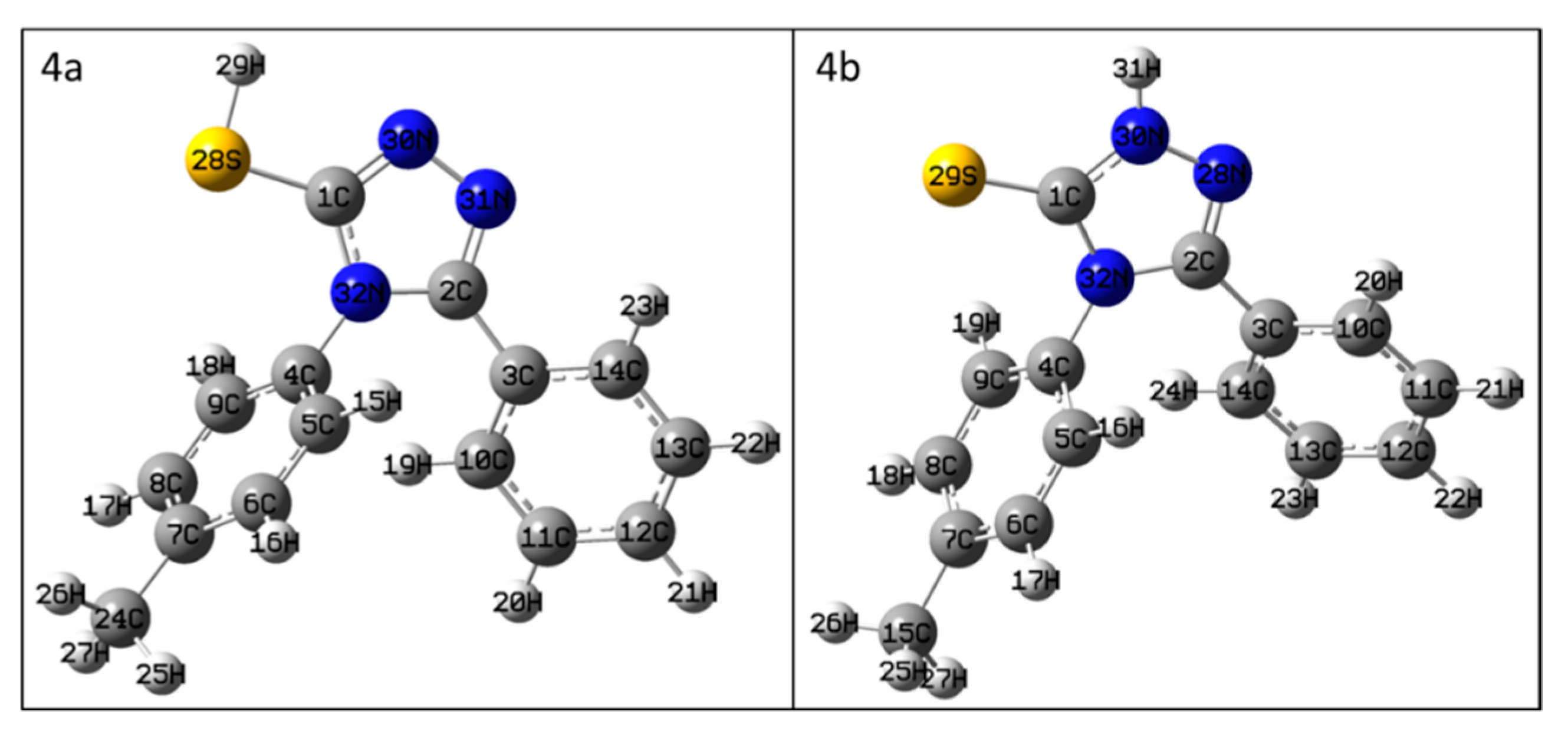
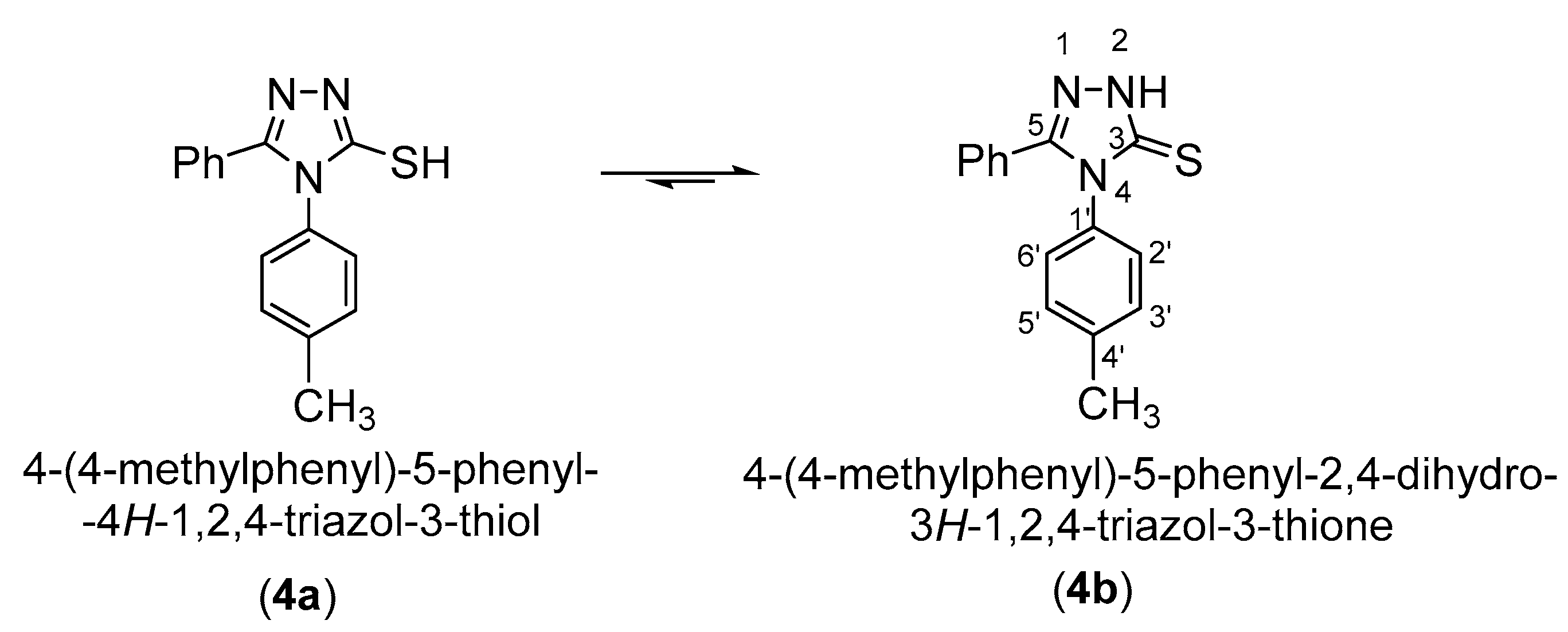
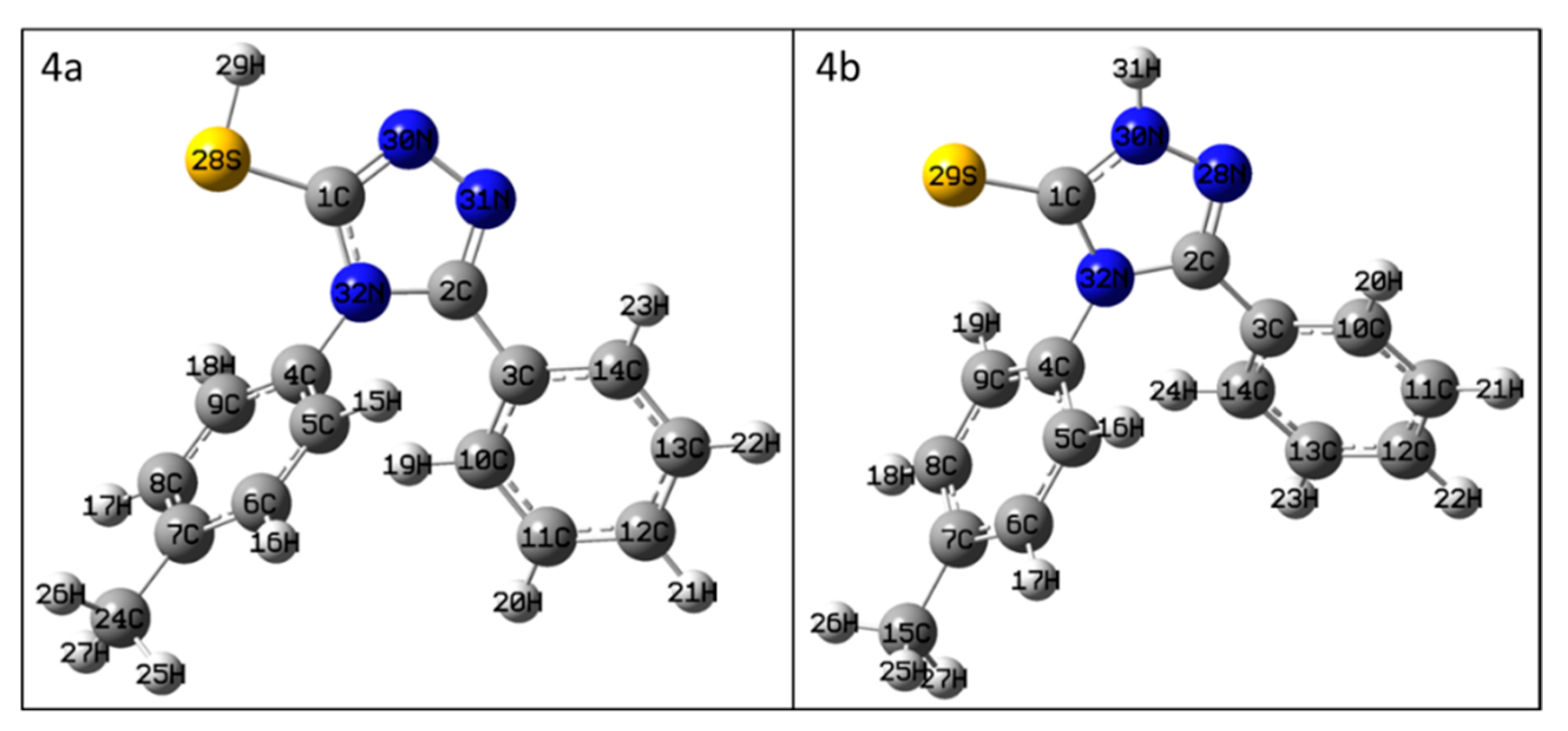
3.1. NMR Characterization of 4-(4-Methylphenyl)-5-phenyl-4H-1,2,4-triazol-3-thione (4)
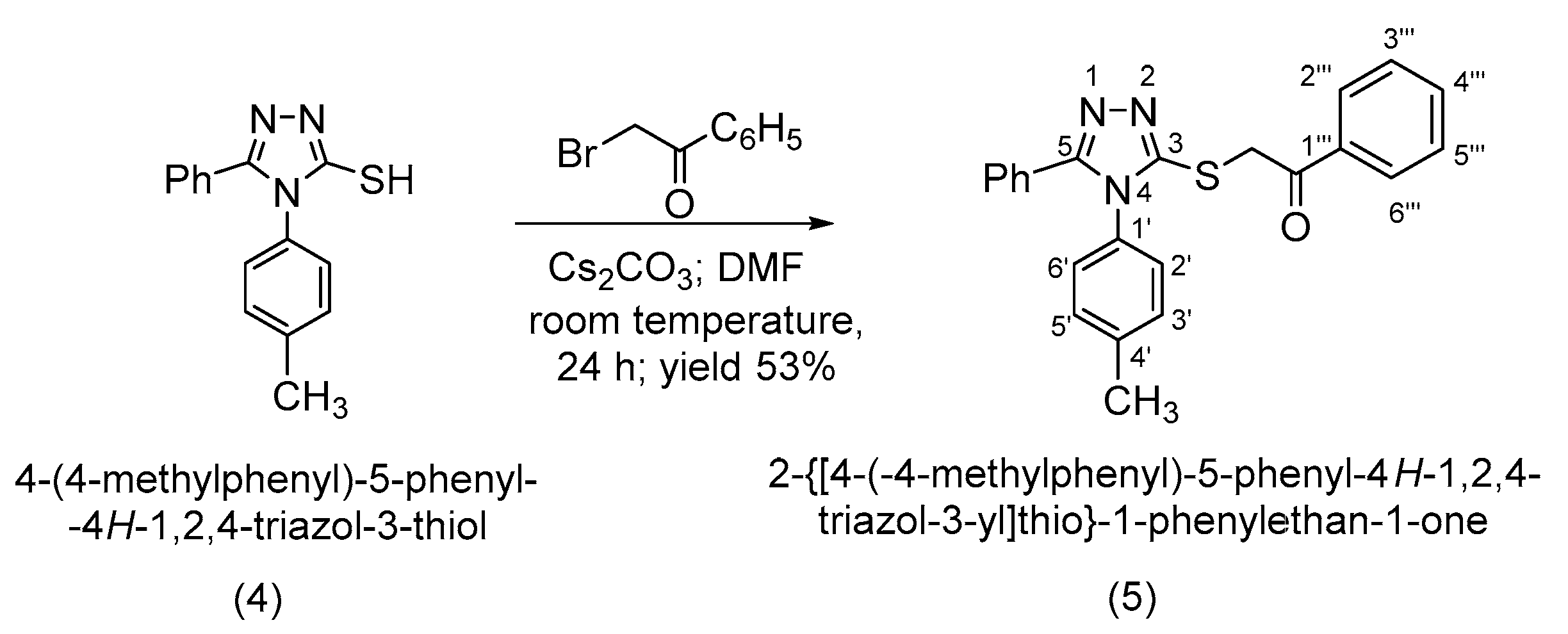
3.2. Synthesis of 2-{[4-(4-Methylphenyl)-5-phenyl-4H-1,2,4-triazol-3-yl]thio}-1-phenylethan-1-one (5)
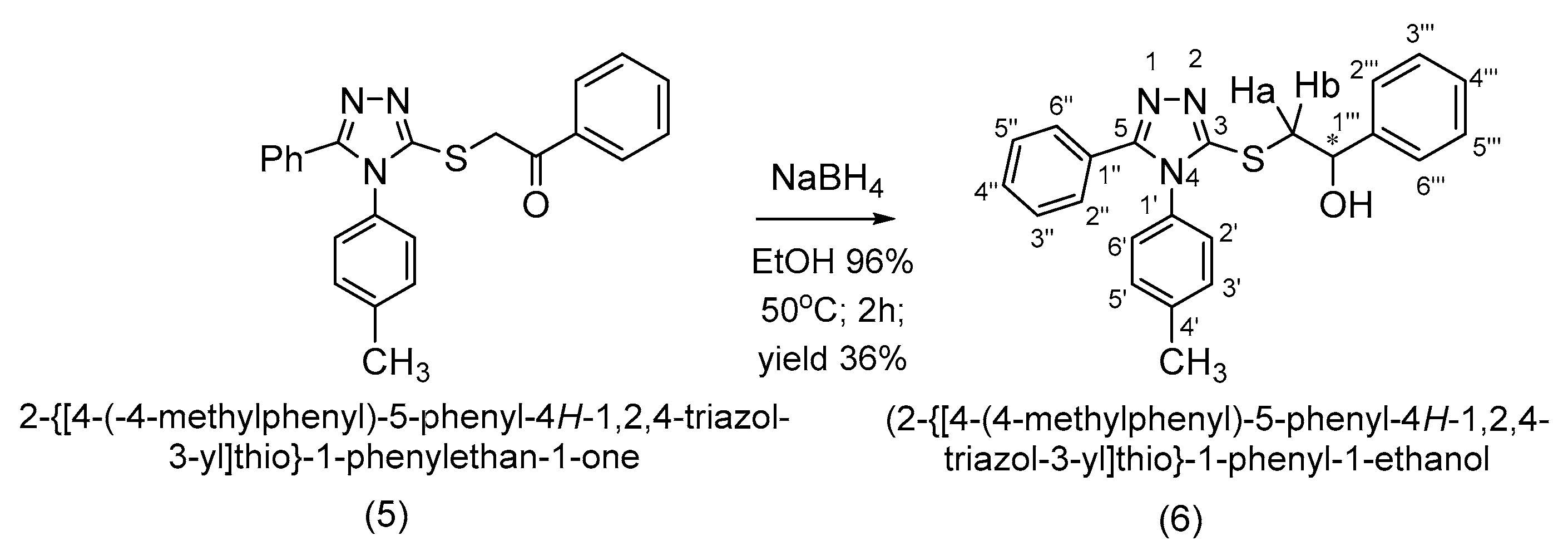
3.3. Synthesis of 2-{[4-(4-Methylphenyl)-5-phenyl-4H-1,2,4-triazol-3-yl]thio}-1-phenyl-1-ethanol (6)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dimri, A.K.; Parmar, S.S. Synthesis of 3-aryl-4-oxothiazolin-2-yl(4-ethoxy-3-methoxy)phenyl hydrazones as possible anticonvulsants. J. Heterocycl. Chem. 1978, 15, 335–336. [Google Scholar] [CrossRef]
- Radl, S. Preparation of some pyrazole derivatives by extrusion of elemental sulfur from 1,3,4-thiadiazines. Collect. Czech. Chem. Commun. 1992, 57, 656–659. [Google Scholar] [CrossRef]
- Nuțiu, M.; Bercean, V.; Birău, M. Synthesis of some 4-aryl-thiosemicarbazides. Ann. West Univ. Timiş. 1996, V, 7–10. [Google Scholar]
- Golovlyova, S.M.; Moskvichev, Y.A.; Alov, E.M.; Kobylinsky, D.B.; Ermolaeva, V.V. Synthesis of novel five-membered nitrogen-containing heterocyclic compounds from derivatives of arylsulfonyl and arylthioacetic and propionic acids. Chem. Heterocycl. Compd. 2001, 37, 1102–1106. [Google Scholar] [CrossRef]
- Ledeți, I.; Bercean, V.; Alexa, A.; Foica, C.; Șuta, L.-M.; Dehelean, C.; Trandafirescu, C.; Muntean, D.; Licker, M.; Fuliaș, A. Preparation and antibacterial properties of substituted 1,2,4-triazoles. J. Chem. 2015, 2015, 879343. [Google Scholar] [CrossRef] [Green Version]
- Salvatore, R.N.; Smith, R.A.; Nischwitz, A.K.; Gavin, T. A mild and highly convenient chemoselective alkylation of thiols using Cs2CO3–TBAI. Tetrahedron Lett. 2005, 46, 8931–8935. [Google Scholar] [CrossRef]
- Varala, R.; Rao, K.S. Cesium salts in organic synthesis: A Review. Curr. Org. Chem. 2015, 19, 1242–1274. [Google Scholar] [CrossRef]
- Gómez, A.B.; Ahlsten, N.; Platero-Prats, A.E.; Martín-Matute, B. Synthesis of 4,5-disubstituted 2-aminothiazoles from α,β-unsaturated ketones: Preparation of 5-benzyl-4methyl-2-aminothiazolium hydrochloride salt. Org. Synth. 2014, 91, 185–200. [Google Scholar] [CrossRef]
- Ahmed, F.F.; Abd El-Hafeez, A.A.; Abbas, S.H.; Abdelhamid, D.; Abdel-Aziz, M. New 1,2,4-triazole-chalcone hybrids induce caspase-3 dependent apoptosis in A549 human lung adenocarcinoma cells. Eur. J. Med. Chem. 2018, 151, 705–722. [Google Scholar] [CrossRef] [PubMed]
- Godhani, D.R.; Sanja, D.B.; Sanghani, A.M. Synthesis and antimicrobial elucidation of [1,2,4]-triazole-3-thione derivatives. J. Chem. Pharm. Res. 2013, 5, 240–243. [Google Scholar]
- Zoumpoulakis, P.; Camoutsis, C.; Pairas, G.; Soković, M.; Glamočlija, J.; Potamitis, C.; Pitsas, A. Synthesis of novel sulfonamide-1,2,4-triazoles, 1,3,4-thiadiazoles and 1,3,4-oxadiazoles, as potential antibacterial and antifungal agents. Bioorg. Med. Chem. 2012, 20, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Jingde, W.; Xinyong, L.; Xianchao, C.; Yuan, C.; Defeng, W.; Zhong, L.; Wenfang, X.; Christophe, P.; Myriam, W.; Erik, D.C. Synthesis of novel derivatives of 4-amino-3-(2-furyl)-5-mercapto-1,2,4-triazole as potential HIV-1 NNRTIs. Molecules 2007, 12, 2003–2016. [Google Scholar]
- Al-Aabdullah, E.S.; Asiri, H.H.; Lahsasni, S.; Habib, E.E.; Ibrahim, T.M.; El-Emam, A.A. Synthesis, antimicrobial, and antiInflammatory activity, of novel S-substituted and N-substituted 5-(1-adamantyl)-1,2,4-triazole-3-thiols. Drug. Des. Dev. Ther. 2014, 8, 505–517. [Google Scholar]
- Maxwell, J.R.; Wasdahl, D.A.; Wolfson, A.C.; Stenberg, V.I. Synthesis of 5-aryl-2H-tetrazoles, 5-aryl-2H-tetrazole-2-acetic acids, and [(4-phenyl-5-aryl-4H-1,2,4-triazol-3-yl)thio]acetic acids as possible superoxide scavengers and antiinflammatory agents. J. Med. Chem. 1984, 27, 1565–1570. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| compound 4a | compound 4b | compound 4 | |||
|---|---|---|---|---|---|
| Atom * | δ (ppm) Calc. | Atom * | δ (ppm) Calc. | Atom * | δ (ppm) Exp. |
| 1C | 141.98 | 1C | 159.79 | 3-C | 168.6 |
| 2C | 144.19 | 2C | 141.09 | 5-C | 150.5 |
| 3C | 116.30 | 3C | 114.86 | 1″-C | 125.8 |
| 4C | 119.74 | 4C | 120.16 | 1′-C | 131.8 |
| 5C | 114.21 | 5C | 114.59 | 2′-C | 128.4 |
| 6C | 117.39 | 6C | 117.04 | 3′-C | 129.7 |
| 7C | 131.63 | 7C | 130.95 | 4′-C | 138.8 |
| 8C | 117.03 | 8C | 116.86 | 5′-C | 129.7 |
| 9C | 115.67 | 9C | 117.70 | 6′-C | 128.4 |
| 10C | 114.25 | 10C | 115.95 | 6″-C | 128.1 |
| 11C | 115.22 | 11C | 115.99 | 5″-C | 128.3 |
| 12C | 115.91 | 12C | 117.34 | 4″-C | 130.2 |
| 13C | 116.15 | 13C | 115.11 | 3″-C | 128.3 |
| 14C | 115.24 | 14C | 115.42 | 2″-C | 128.1 |
| 15H | 8.10 | 15C | 12.81 | -CH3 | 20.7 |
| 16H | 8.54 | 16H | 7.27 | 2′-H | 7.22 |
| 17H | 8.82 | 17H | 7.74 | 3′-H | 7.28 |
| 18H | 8.76 | 18H | 8.09 | 5′-H | 7.28 |
| 19H | 8.08 | 19H | 7.97 | 6′-H | 7.22 |
| 20H | 8.38 | 20H | 8.37 | 6″-H | 7.36−7.31 |
| 21H | 8.60 | 21H | 7.98 | 3″-H | 7.36−7.31 |
| 22H | 8.68 | 22H | 7.92 | 4″-H | 7.43−7.40 |
| 23H | 9.24 | 23H | 7.65 | 5″-H | 7.36−7.31 |
| 24C | 12.69 | 24H | 7.47 | 2″-H | 7.36−7.31 |
| 25H | 3.36 | 25H | 2.49 | -CH3 | 2.35 |
| 26H | 3.65 | 26H | 2.72 | -CH3 | 2.35 |
| 27H | 3.29 | 27H | 2.90 | -CH3 | 2.35 |
| 28S | - | 28N | 270.52 | - | - |
| 29H | 5.86 | 29S | - | - | - |
| 30N | 300.94 | 30N | 196.97 | - | - |
| 31N | 315.84 | 31H | 9.89 | -N-H | 14.11 |
| 32N | 184.26 | 32N | 190.79 | 4-N | 183.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valicsek, V.-S.; Badea, V. (R,S)-2-{[4-(4-Methylphenyl)-5-phenyl-4H-1,2,4-triazol-3-yl]thio}-1-phenyl-1-ethanol. Molbank 2021, 2021, M1241. https://doi.org/10.3390/M1241
Valicsek V-S, Badea V. (R,S)-2-{[4-(4-Methylphenyl)-5-phenyl-4H-1,2,4-triazol-3-yl]thio}-1-phenyl-1-ethanol. Molbank. 2021; 2021(3):M1241. https://doi.org/10.3390/M1241
Chicago/Turabian StyleValicsek, Vladislav-Silvestru, and Valentin Badea. 2021. "(R,S)-2-{[4-(4-Methylphenyl)-5-phenyl-4H-1,2,4-triazol-3-yl]thio}-1-phenyl-1-ethanol" Molbank 2021, no. 3: M1241. https://doi.org/10.3390/M1241
APA StyleValicsek, V.-S., & Badea, V. (2021). (R,S)-2-{[4-(4-Methylphenyl)-5-phenyl-4H-1,2,4-triazol-3-yl]thio}-1-phenyl-1-ethanol. Molbank, 2021(3), M1241. https://doi.org/10.3390/M1241