
Diethyl (2-(4-Phenyl-1H-1,2,3-triazol-1-yl)benzyl) Phosphate


Abstract
1. Introduction
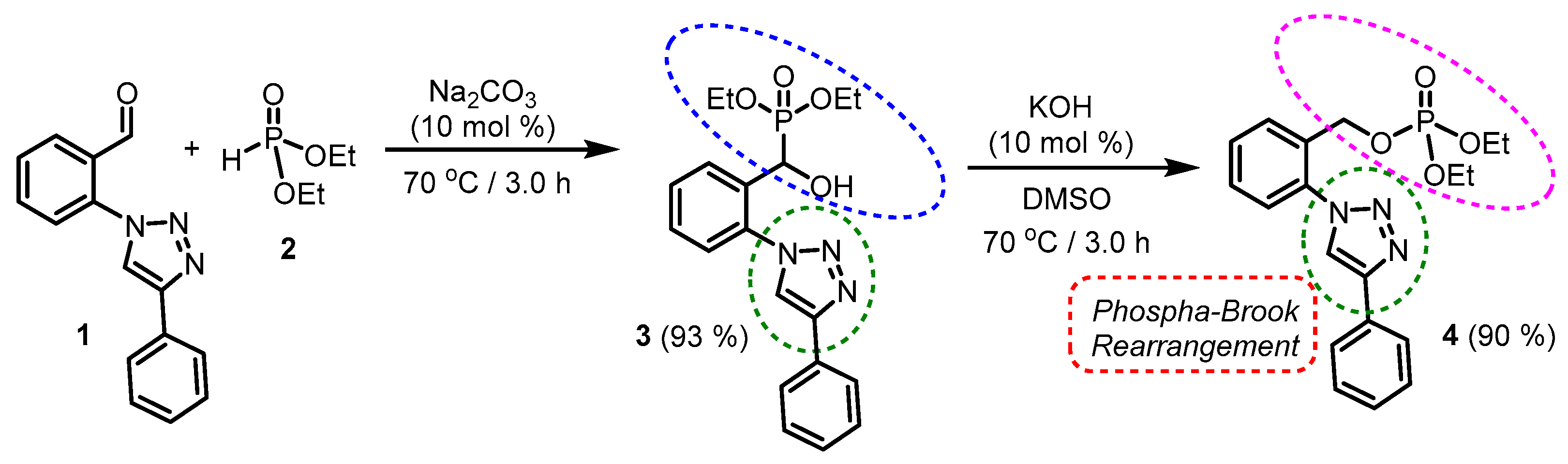
2. Results
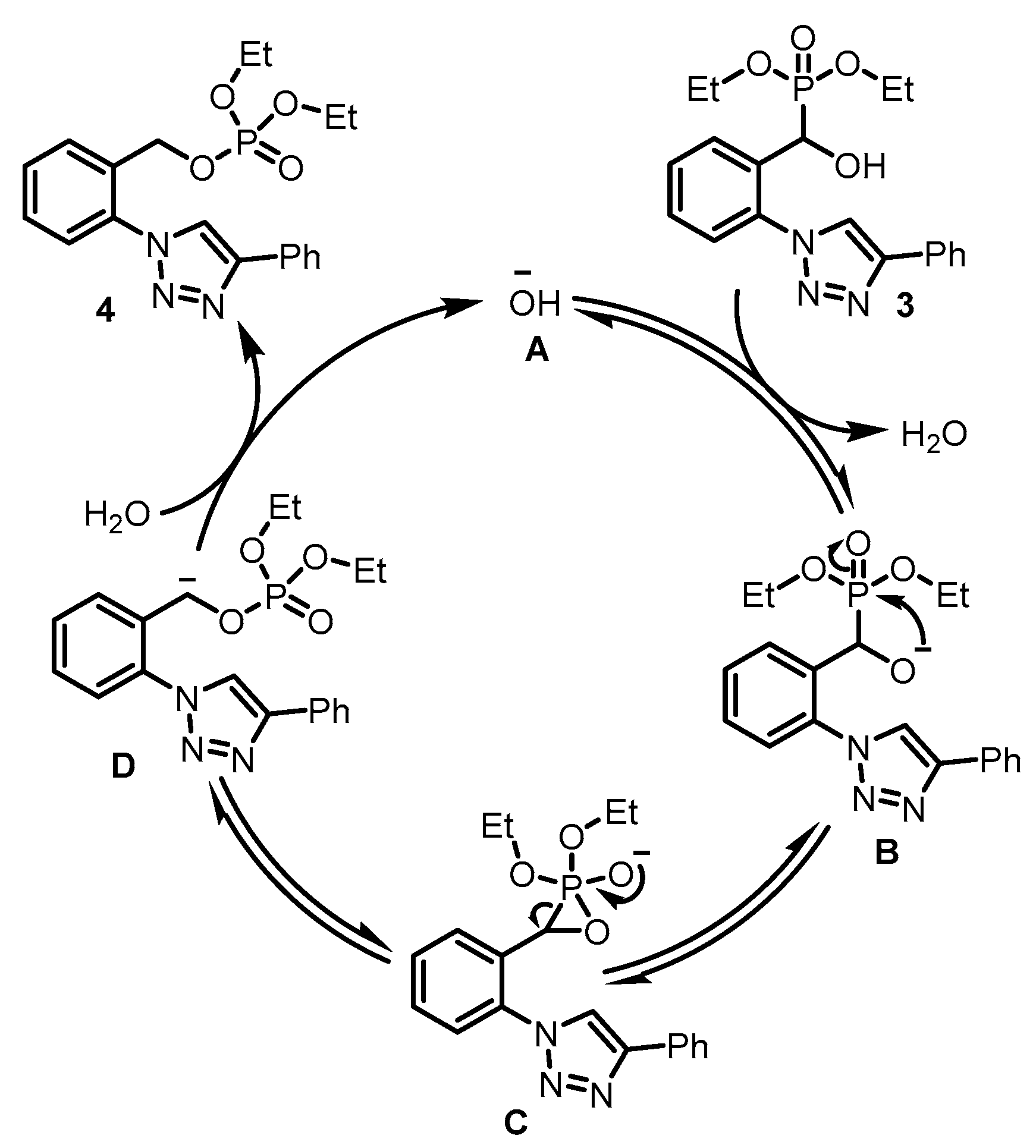
3. Discussion
4. Materials and Methods
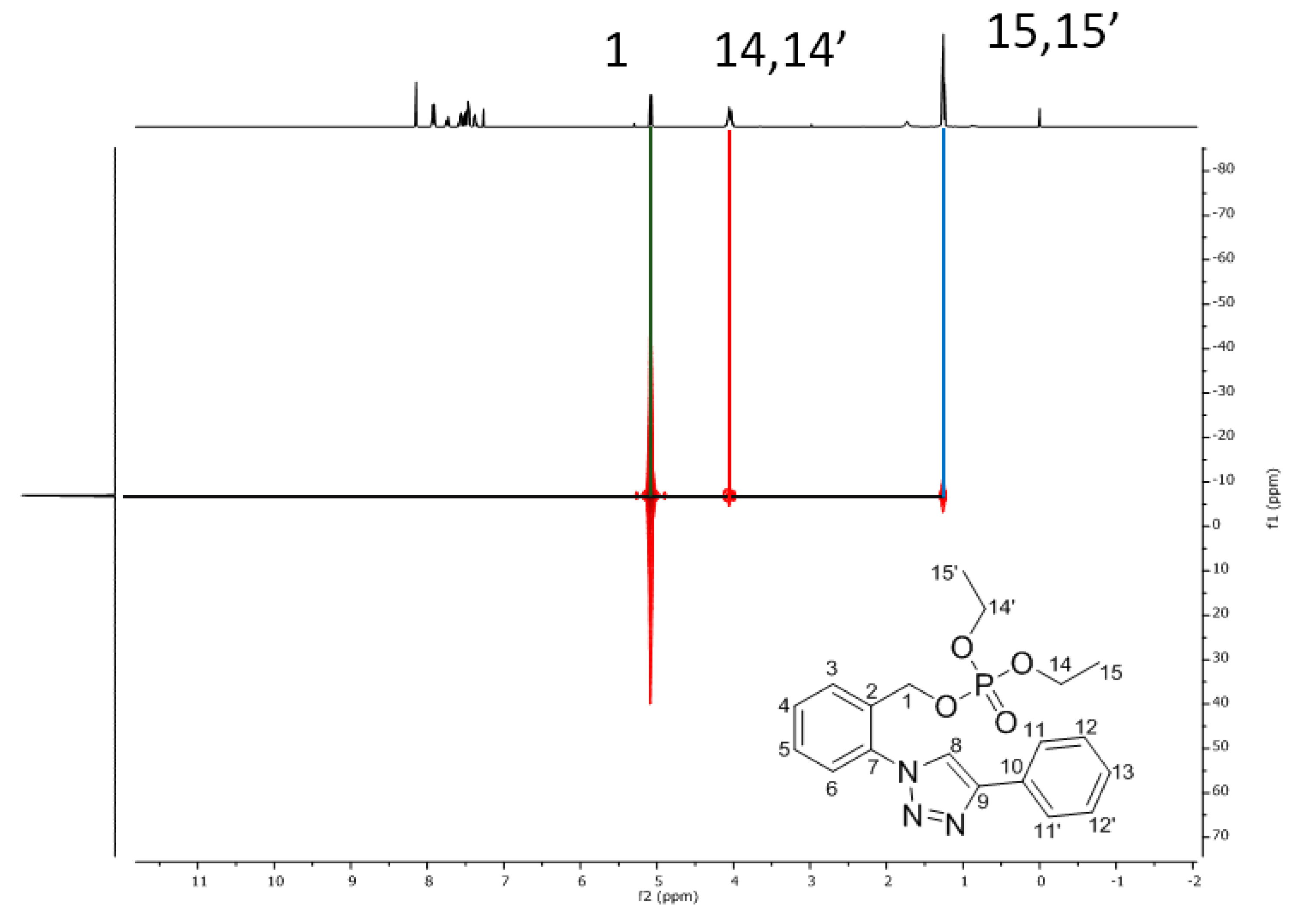
Spectral Data of Compounds 3 and 4
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eastman, R.T.; Roth, J.S.; Brimacombe, K.R.; Simeonov, A.; Shen, M.; Patnaik, S.; Hall, M.D. Remdesivir: A Review of Its Discovery and Development Leading to Emergency Use Authorization for Treatment of COVID-19. ACS Cen. Sci. 2020, 6, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Haji, M. Multicomponent reactions: A simple and efficient route to heterocyclic phosphonates. Beilstein J. Org. Chem. 2016, 12, 1269–1301. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Sello, G.; Sisti, M. Aminophosphonic acids and derivatives. Synthesis and biological applications. Curr. Med. Chem. 2010, 17, 264–289. [Google Scholar] [CrossRef] [PubMed]
- Naydenova, E.D.; Todorov, P.T.; Troev, K.D. Recent synthesis of aminophosphonic acids as potential biological importance. Amino Acids 2010, 38, 23–30. [Google Scholar] [CrossRef]
- Murphy, P.J. Organophosphorus Reagents; Oxford University Press: Oxford, UK, 2004. [Google Scholar]
- Quin, L.D. A Guide to Organophosphorus Chemistry; Wiley: New York, NY, USA, 2000. [Google Scholar]
- Gardner, G.; Steffens, J.J.; Grayson, B.T.; Kleier, D.A. 2-Methylcinnolinium Herbicides: Effect of 2-Methylcinnolinium-4-(O-methyl phosphonate) on Photosynthetic Electron Transport. J. Agric. Food Chem. 1992, 40, 318–321. [Google Scholar] [CrossRef]
- Olszewski, T.K. Environmentally Benign Syntheses of α-Substituted Phosphonates: Preparation of α-Amino- and α-Hydroxyphosphonates in Water, in Ionic Liquids, and under Solvent-Free Conditions. Synthesis 2014, 46, 403–429. [Google Scholar] [CrossRef]
- Santacroce, V.; Paris, E.; Cauzzi, D.; Maggi, R.; Maestri, G. A Simple Heterogeneous Catalyst for Phosphite Addition on Carbonyl Groups. Eur. J. Org. Chem. 2016, 463–466. [Google Scholar] [CrossRef]
- Vamisetti, G.B.; Chowdhury, R.; Chosh, S.K. Organocatalytic decarboxylative aldol reaction of β-ketoacids with α-ketophosphonates en route to the enantioselective synthesis of tertiary α-hydroxyphosphonates. Org. Biomol. Chem. 2017, 15, 3869–3873. [Google Scholar] [CrossRef]
- Fischer, H.C.; Prost, L.; Montchamp, J.L. Organophosphorus Chemistry without PCl3: A Bridge from Hypophosphorous Acid to H-Phosphonate Diesters. Eur. J. Org. Chem. 2013, 2013, 7973–7978. [Google Scholar] [CrossRef]
- Montchamp, J.L. Phosphinate Chemistry in the 21st Century: A Viable Alternative to the Use of Phosphorus Trichloride in Organophosphorus Synthesis. Acc. Chem. Res. 2014, 47, 77–87. [Google Scholar] [CrossRef]
- Saha, D.; Kaur, T.; Singh, N.; Singh, U.P.; Sharma, A. Rapid Access to New Thiazepinyl and Oxazepinyl Phosphonates through a Green Pudovik Reaction. Asian J. Org. Chem. 2016, 5, 82–90. [Google Scholar] [CrossRef]
- Janesko, B.G.; Fisher, H.C.; Bridle, M.J.; Montchamp, J.-L. P(=O)H to P–OH Tautomerism: A Theoretical and Experimental Study. J. Org. Chem. 2015, 80, 10025–10032. [Google Scholar] [CrossRef] [PubMed]
- Vincze, D.; Ábrányi-Balogh, P.; Bagi, P.; Keglevich, G. A Mechanistic Study on the Tautomerism of H-Phosphonates, H-Phosphinates and Secondary Phosphine Oxides. Molecules 2019, 24, 3859. [Google Scholar] [CrossRef] [PubMed]
- Pallikonda, G.; Chakravarty, M.; Sahoo, M.K. An easy access to α-aryl substituted γ-ketophosphonates: Lewis acid mediated reactions of 1,3-diketones with α-hydroxyphosphonates and tandem regioselective C–C bond cleavage. Org. Biomol. Chem. 2014, 12, 7140–7149. [Google Scholar] [CrossRef]
- Prechelmacher, S.; Mereiter, K.; Hammerschmidt, F. The α-hydroxyphosphonate-phosphate rearrangement of a noncyclic substrate–some new observations. Org. Biomol. Chem. 2018, 16, 3672–3680. [Google Scholar] [CrossRef]
- Lopez, Ó.; Fernadez-Bolañosa, J.G.; Gil, M.V. New trends in pest control: The search for greener insecticides. Green Chem. 2005, 7, 431–442. [Google Scholar] [CrossRef]
- Kasthuri, M.; Chaloin, L.; Périgaud, C.; Peyrottes, S. Synthesis of (R)- and (S)-β-hydroxyphosphonate acyclonucleosides: Structural analogues of Adefovir (PMEA). Tetrahedron Asymmetry 2011, 22, 1505–1511. [Google Scholar] [CrossRef]
- Pokalwar, R.U.; Hangarge, R.V.; Maske, P.V.; Shingare, M.S. Synthesis and antibacterial activities of α-hydroxyphosphonates and α-acetyloxyphosphonates derived from 2-chloroquinoline-3-carbaldehyde. ARKIVOC 2006, 11, 196–204. [Google Scholar] [CrossRef]
- Hecker, S.J.; Erion, M.D. Prodrugs of Phosphates and Phosphonates. J. Med. Chem. 2008, 51, 2328–2345. [Google Scholar] [CrossRef]
- Bowler, M.W.; Cliff, M.J.; Waltho, J.P.; Blackburn, G.M. Why did Nature select phosphate for its dominant roles in biology? New J. Chem. 2010, 34, 784–794. [Google Scholar] [CrossRef]
- Jessen, H.J. The Hitchhiker’s Guide to Organophosphate Chemistry. Synlett 2018, 29, 699–713. [Google Scholar] [CrossRef]
- Lima, Y.R.; Da Costa, G.P.; Xavier, M.C.D.F.; De Moraes, M.C.; Barcellos, T.; Alves, D.; Silva, M.S. Synthesis of α-Hydroxyphosphonates Containing Functionalized 1,2,3-Triazoles. ChemistrySelect 2020, 5, 12487–12493. [Google Scholar] [CrossRef]
- Alves, D.; Goldani, B.; Lenardão, E.J.; Perin, G.; Schumacher, R.F.; Paixão, M.W. Copper Catalysis and Organocatalysis Showing the Way: Synthesis of Selenium-Containing Highly Functionalized 1,2,3-Triazoles. Chem. Rec. 2018, 18, 527–542. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.F.; Costa, G.P.; Luque, R.; Alves, D.; Fajardo, A.R. Polysaccharide-based superporous hydrogel embedded with copper nanoparticles: A green and versatile catalyst for the synthesis of 1,2,3-triazoles. Catal. Sci. Technol. 2019, 9, 136–145. [Google Scholar] [CrossRef]
- Costa, G.P.; Baldinotti, R.S.M.; Fronza, M.G.; Nascimento, J.E.R.; Dias, Í.F.C.; Sonego, M.S.; Seixas, F.K.; Collares, T.; Perin, G.; Jacob, R.G.; et al. Synthesis, Molecular Docking, and Preliminary Evaluation of 2-(1,2,3-Triazoyl)benzaldehydes As Multifunctional Agents for the Treatment of Alzheimer’s Disease. ChemMedChem 2020, 15, 610–622. [Google Scholar] [CrossRef]
- Costa, G.P.; Bach, M.; Moraes, M.; Silva, T.B.; Lenardão, E.J.; Silva, M.S.; Alves, D. Sequential Organocatalytic Synthesis of [1,2,3] triazoyl [1,5a] quinolines. Adv. Synth. Catal. 2020, 362, 5044–5055. [Google Scholar] [CrossRef]
- Tian, X.; Yang, F.; Rasina, D.; Bauer, M.; Warratz, S.; Ferlin, F.; Vaccaro, L.; Ackerman, L. C–H arylations of 1,2,3-triazoles by reusable heterogeneous palladium catalysts in biomass-derived γ-valerolactone. Chem. Commun. 2016, 52, 9777–9780. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sharma, B.; Mehra, V.; Kumar, V. Recent accomplishments on the synthetic/biological facets of pharmacologically active 1H-1,2,3-triazoles. Eur. J. Med. Chem. 2021, 212, 113069–113159. [Google Scholar] [CrossRef] [PubMed]
- Pallitsch, K.; Roller, A.; Hammerschmidt, F. The Stereochemical Course of the α-Hydroxyphosphonate–Phosphate Rearrangement. Chem. Eur. J. 2015, 21, 10200–10206. [Google Scholar] [CrossRef] [PubMed]
- Rádai, Z.; Szabó, R.; Szigetvári, A.; Kiss, N.Z.; Mucsi, Z.; Keglevich, G. A Study on the Rearrangement of Dialkyl 1-Aryl-1-hydroxymethylphosphonates to Benzyl Phosphates. Curr. Org. Chem. 2020, 24, 465–471. [Google Scholar] [CrossRef]
- Kim, T.-W.; Islam, T.; Jung, K.-Y. Design and synthesis of non-steroidal diclofenac derivatives as anti-inflammatory drugs. J. Ind. Eng. Chem. 2010, 16, 461–466. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Number | 1H (ppm) | 13C (ppm) | 13C HMBC |
| 1 | 5.08 (d, JH-P = 16.5 Hz) | 65.0 (d, JC-P = 5.1 Hz) | C3, C7 |
| 2 | --- | 131.5 (d, JC-P = 7.6 Hz) | --- |
| 3 | 7.74 (d, JH-H = 7.0 Hz) | 130.5 | C5, C7 |
| 4 | 7.60–7.54 (m) | 130.2 | C2, C6 |
| 5 | 7.60–7.54 (m) | 129.8 | C3, C7 |
| 6 | 7.53–7.51 (m) | 125.9 | C2, C4 |
| 7 | --- | 135.9 | --- |
| 8 | 8.15 (s) | 121.6 | C9 |
| 9 | --- | 148.1 | --- |
| 10 | --- | 130.2 | --- |
| 11, 11′ | 7.92 (d, JH-H = 8.0 Hz) | 126.0 | C8, C9, C12 |
| 12, 12′ | 7.49–7.45 (m) | 129.1 | C10, C11, C11′, C13 |
| 13 | 7.40–7.36 (m) | 128.6 | C11 |
| 14, 14′ | 4.12–3.99 (m) | 64.2 (d, JC-P = 5.8 Hz) | C15, C15′ |
| 15, 15′ | 1.25 (t, JH-H = 7.1 Hz) | 16.2 (d, JC-P = 6.6 Hz) | C14, C14′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, G.P.d.; Alves, D.; Silva, M.S. Diethyl (2-(4-Phenyl-1H-1,2,3-triazol-1-yl)benzyl) Phosphate. Molbank 2021, 2021, M1223. https://doi.org/10.3390/M1223
Costa GPd, Alves D, Silva MS. Diethyl (2-(4-Phenyl-1H-1,2,3-triazol-1-yl)benzyl) Phosphate. Molbank. 2021; 2021(2):M1223. https://doi.org/10.3390/M1223
Chicago/Turabian StyleCosta, Gabriel P. da, Diego Alves, and Márcio S. Silva. 2021. "Diethyl (2-(4-Phenyl-1H-1,2,3-triazol-1-yl)benzyl) Phosphate" Molbank 2021, no. 2: M1223. https://doi.org/10.3390/M1223
APA StyleCosta, G. P. d., Alves, D., & Silva, M. S. (2021). Diethyl (2-(4-Phenyl-1H-1,2,3-triazol-1-yl)benzyl) Phosphate. Molbank, 2021(2), M1223. https://doi.org/10.3390/M1223