
(E)-4-(3-(3-(4-Methoxyphenyl)acryloyl)phenoxy)butyl 2-Hydroxybenzoate



 and
and
Abstract
1. Introduction
2. Results and Discussions
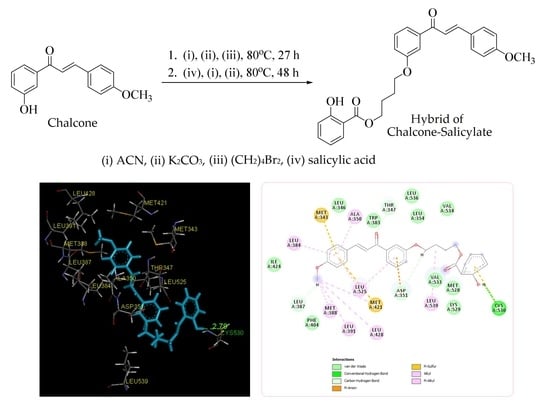
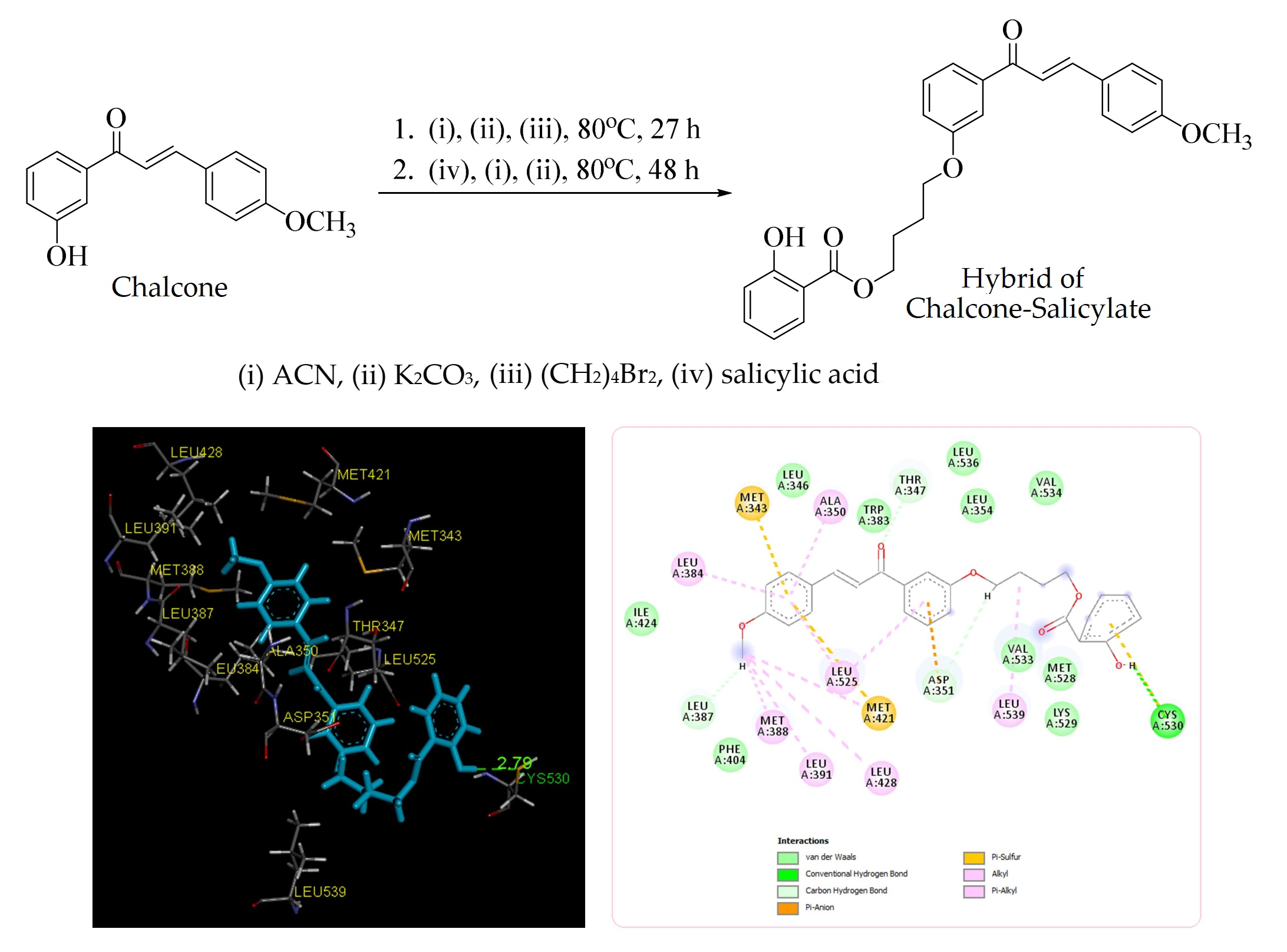
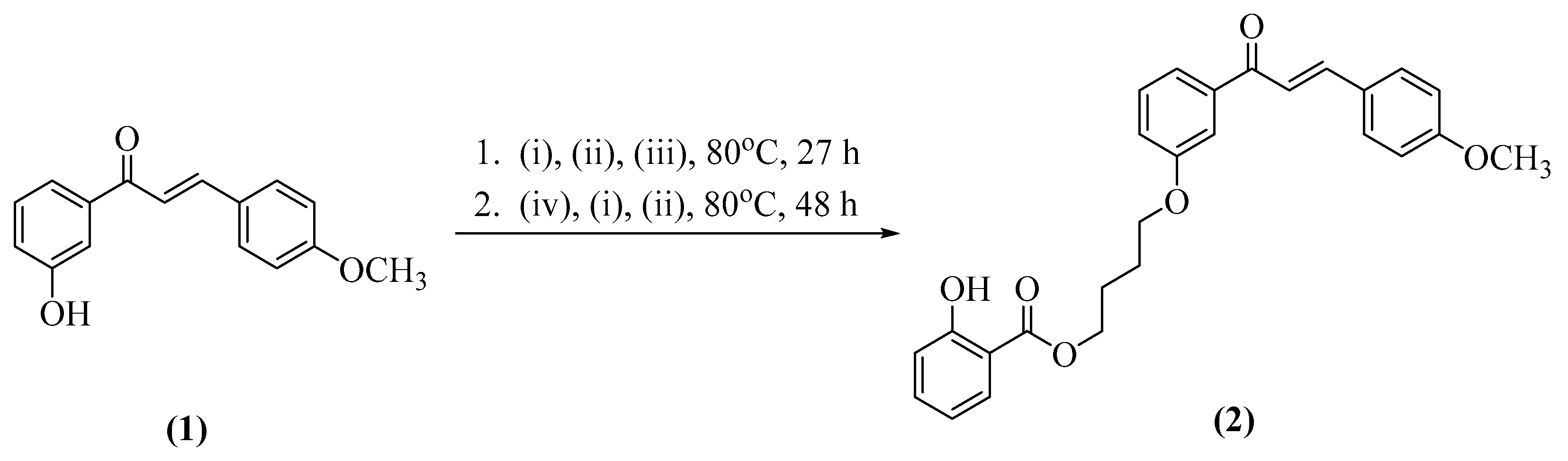
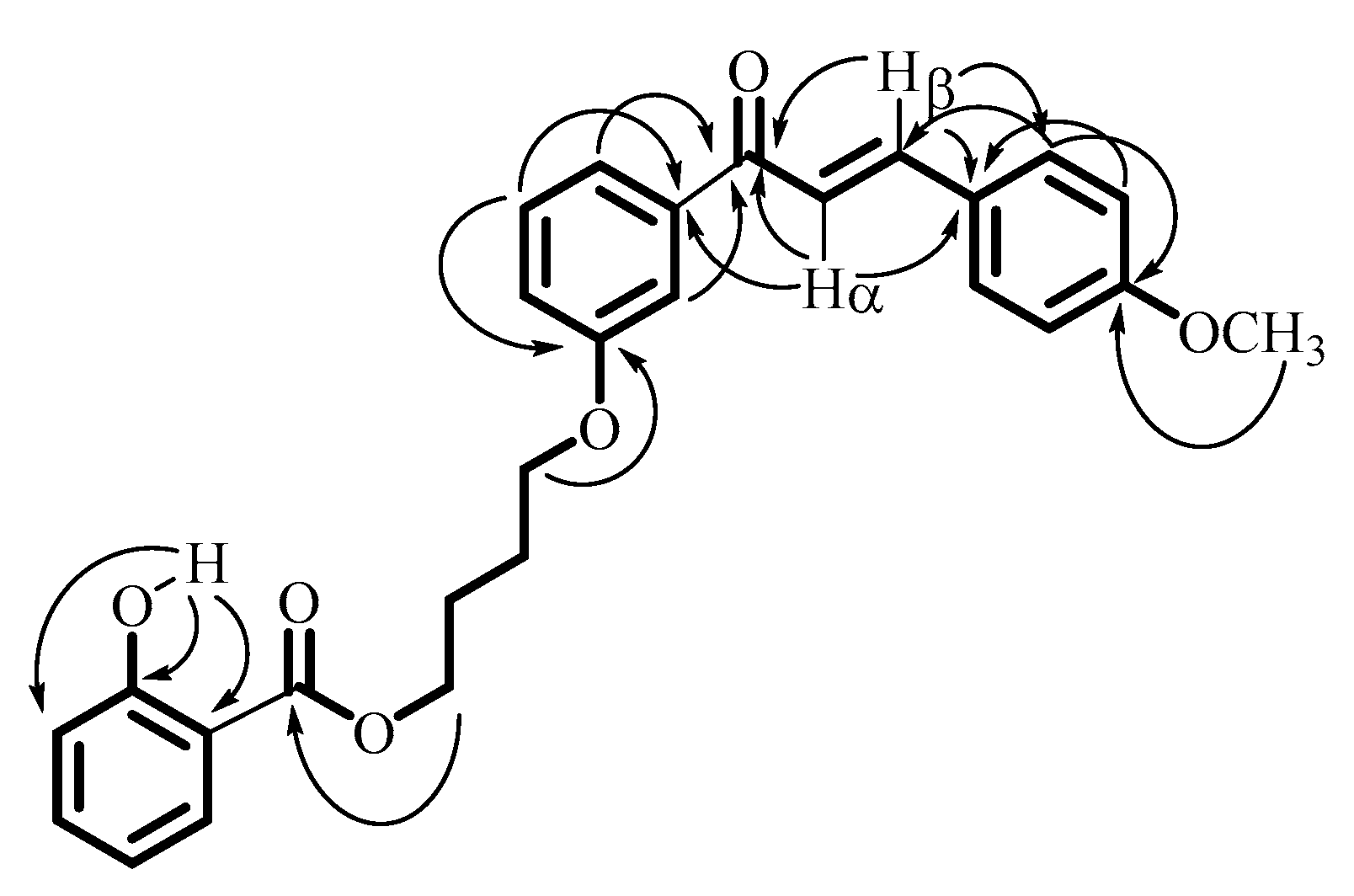
2.1. Synthesis of Title Compound (2)
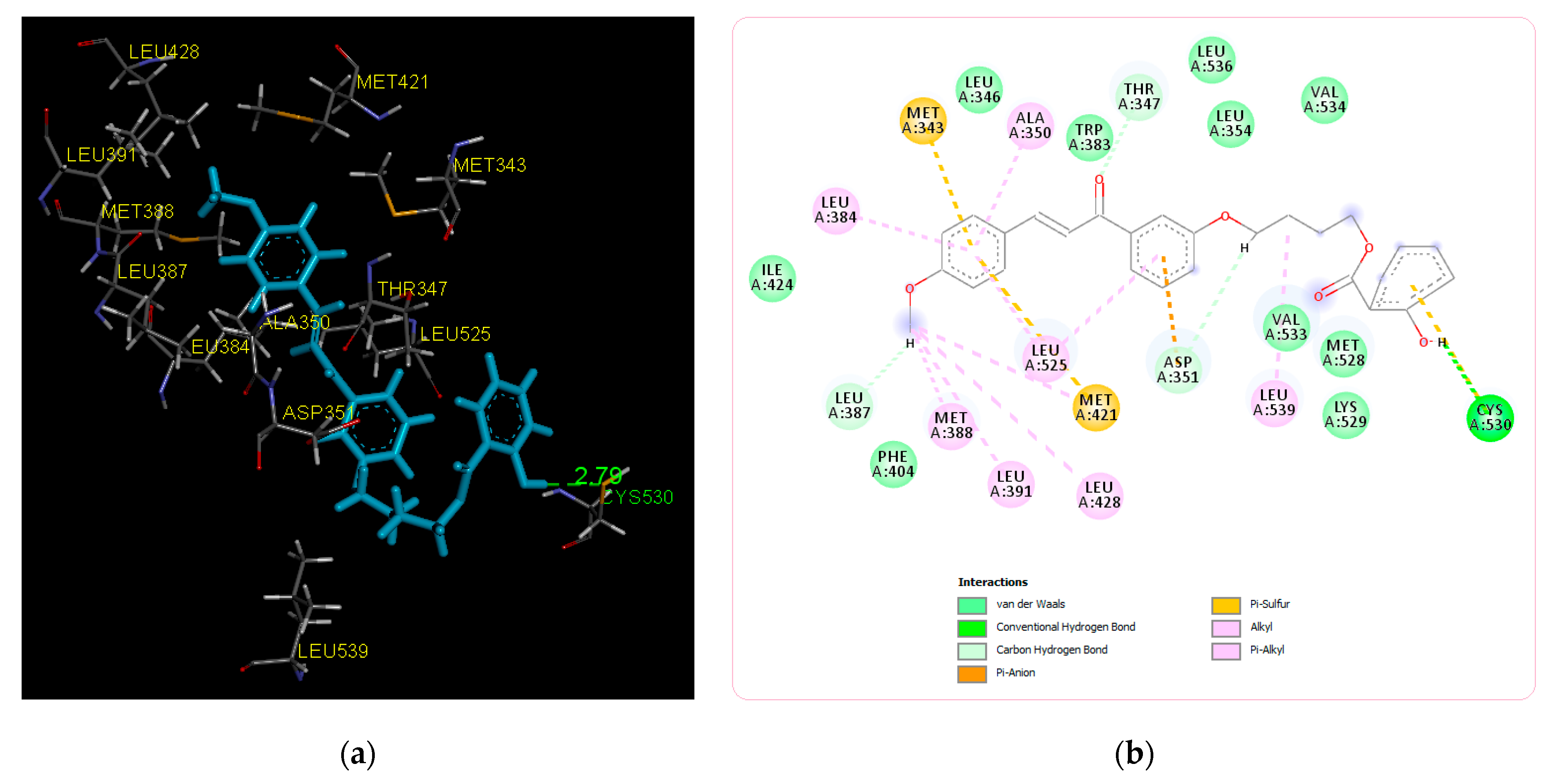
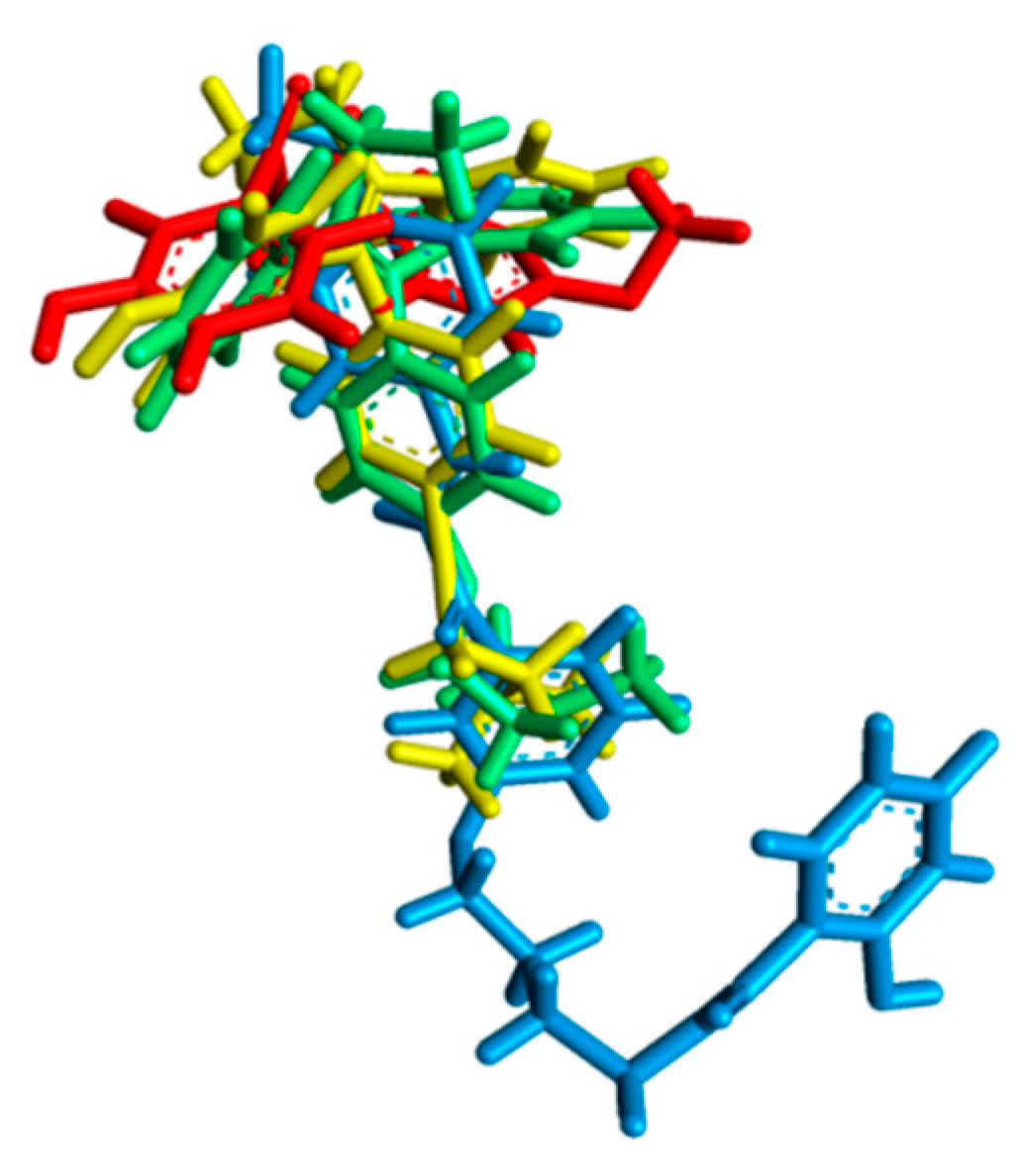
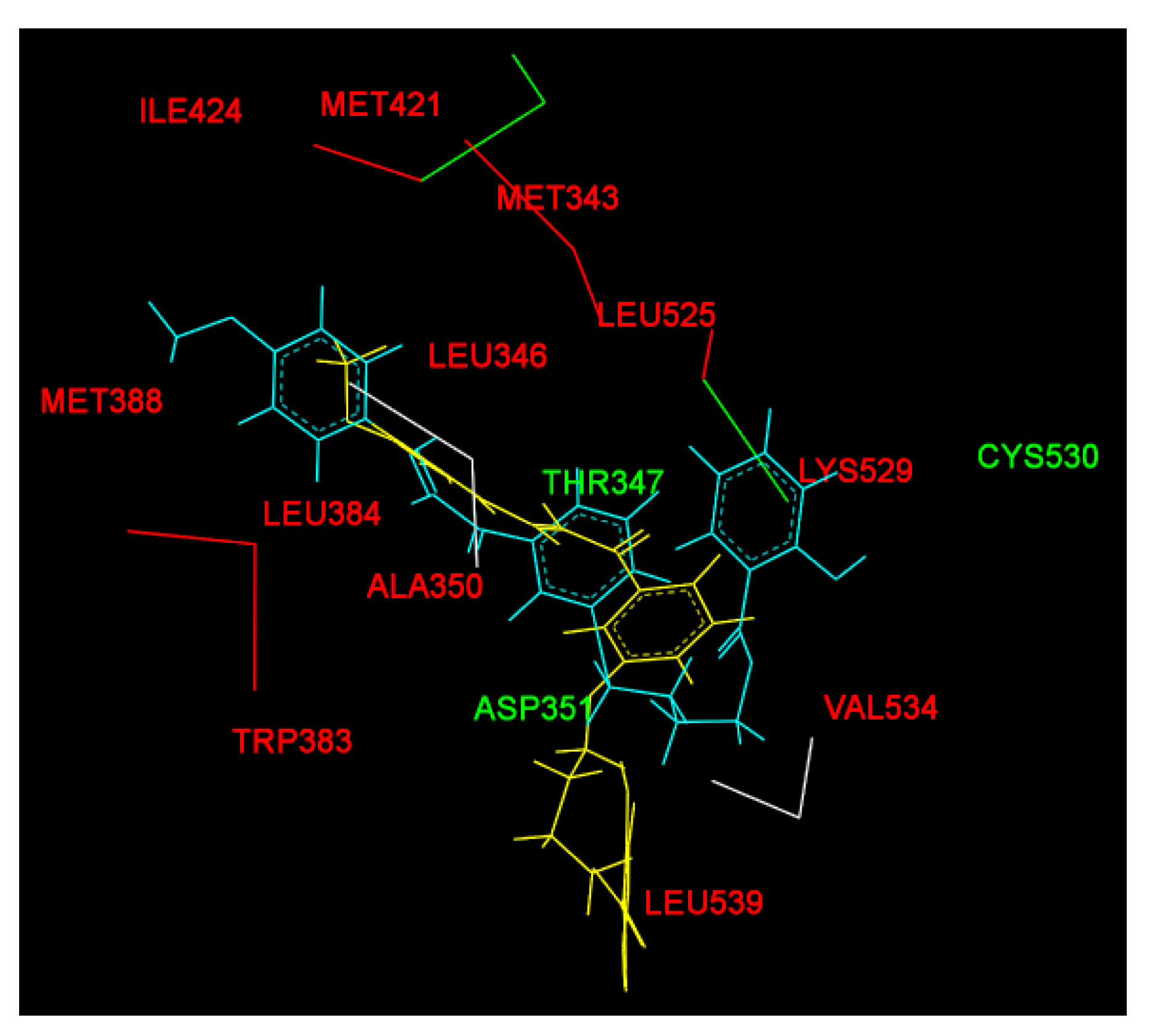
2.2. Molecular Docking Study and MD Simulation
3. Materials and Methods
3.1. Materials
3.2. Instrumentations
3.3. Methods
3.3.1. Synthesis of (E)-4-(3-(3-(4-Methoxyphenyl)acryloyl)phenoxy)butyl 2-Hydroxybenzoate
3.3.2. Molecular Docking Study
3.3.3. MD Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
| UV-Vis | Ultraviolet-Visible |
| FT-IR | Fourier Transform-Infra Red |
| HRMS | High Resolution Mass Spectroscopy |
| 1D NMR | 1 Dimensional Nuclear Magnetic Resonance |
| 2D NMR | 2 Dimensional Nuclear Magnetic Resonance |
| COSY | Correlation of Spectroscopy |
| TOCSY | Total Correlated Spectroscopy |
| HSQC | Heteronuclear Single Quantum Correlation |
| HMBC | Heteronuclear Multiple Bond Correlation |
| ERα | Estrogen Receptor Alpha |
| RMSD | Root Mean Square Deviation |
| MD NPA CHARMM27 | Molecular Dynamic Nosé-Poincaré-Andersen Chemistry at Harvard Macromolecular Mechanics |
| 4-OHT | 4-Hydroxy Tamoxifen |
References
- Zamri, A.; Teruna, H.Y.; Ikhtiarudin, I. The influence of power variations on selectivity of 2′-hydroxychalcone analogue under microwave irradiation. Molekul 2016, 11, 299–307. [Google Scholar] [CrossRef]
- Ikhtiarudina, I.; Agistia, N.; Frimayanti, N.; Harlianti, T.; Jasril, J. Microwave-assisted synthesis of 1-(4-hydroxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one and its activities as an antioxidant, sunscreen, and antibacterial. J. Kim. Sains Dan Apl. 2020, 23, 51–60. [Google Scholar] [CrossRef]
- Ikhtiarudin, I.; Agistia, N.; Harlianti, T.; Zamri, A. Sintesis dan potensi aktivitas tabir surya senyawa analog kalkon turunan 3′-hidroksiasetofenon dan 4-metoksibenzaldehid. J. Photon 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Syam, S.; Abdelwahab, S.I.; Al-Mamary, M.A.; Mohan, S. Synthesis of chalcones with anticancer activities. Molecules 2012, 17, 6179–6195. [Google Scholar] [CrossRef]
- Dona, R.; Frimayanti, N.; Ikhtiarudin, I.; Iskandar, B.; Maulana, F.; Silalahi, N.T. In silico, synthesis and cytotoxic activity of p-methoxy chalcone on human breast cancer MCF-7 cell line. J. Sains Farm. Klin. 2019, 6, 243–249. [Google Scholar] [CrossRef]
- Xiong, R.; He, D.; Deng, X.; Liu, J.; Lei, X.; Xie, Z.; Cao, X.; Chen, Y.; Peng, J.; Tang, G. Design, synthesis and biological evaluation of tryptamine salicylic acid derivatives as potential antitumor agents. Med. Chem. Commun. 2019, 10, 573–583. [Google Scholar] [CrossRef]
- Xu, Q.B.; Chen, X.F.; Feng, J.; Miao, J.F.; Liu, J.; Liu, F.T.; Niu, B.X.; Cai, J.Y.; Huang, C.; Zhang, Y.; et al. Design, synthesis and biological evaluation of hybrids of β-carboline and salicylic acid as potential anticancer and apoptosis inducing agents. Sci. Rep. 2016, 6, 36238. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Chang, R.-A.; Huang, H.-Y.; Wang, X.-M.; Wang, X.-Y.; Chen, L.; Ling, Y. Synthesis and biological evaluation of novel farnesylthiosalicylic acid/salicylic acid hybrids as potential anti-tumor agents. Chin. Chem. Lett. 2014, 25, 1545–1549. [Google Scholar] [CrossRef]
- Pawełczyk, A.; Sowa-Kasprzak, K.; Olender, D.; Zaprutko, L. Molecular consortia—Various structural and synthetic concepts for more effective therapeutics synthesis. Int. J. Mol. Sci. 2018, 19, 1104. [Google Scholar] [CrossRef] [PubMed]
- Shchepinov, M.S.; Case-Green, S.C.; Southern, E.M. Steric factors influencing hybridisation of nucleic acids to oligonucleotide arrays. Nucleic Acids Res. 1997, 25, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Urgel, J.I.; Giovannantonio, M.D.; Gandus, G.; Chen, Q.; Liu, X.; Hayashi, H.; Ruffieux, P.; Decurtins, S.; Narita, A.; Passerone, D.; et al. Overcoming steric hindrance in aryl-aryl homocoupling via on-surface copolymerization. ChemPhysChem 2019, 20, 2360–2366. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Partridge, A.; Wu, Y. Improving nanoparticle-enhanced surface plasmon resonance detection of small molecules by reducing steric hindrance via molecular linkers. Talanta 2019, 198, 350–357. [Google Scholar] [CrossRef]
- Burns, K.A.; Korach, K.S. Estrogen receptors and human disease: An update. Arch. Toxicol. 2012, 86, 1491–1504. [Google Scholar] [CrossRef]
- Livezey, M.; Kim, J.E.; Shapiro, D.J. A new role for estrogen receptor α in cell proliferation and cancer: Activating the anticipatory unfolded protein response. Front. Endocrinol. 2018, 9, 325. [Google Scholar] [CrossRef]
- Feng, Y.; Spezia, M.; Huang, S.; Yuan, C.; Zeng, Z.; Zhang, L.; Ji, X.; Liu, W.; Huang, B.; Luo, W.; et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis. 2018, 5, 77–106. [Google Scholar] [CrossRef]
- Nadji, M.; Gomez-Fernandez, C.; Ganjei-Azar, P.; Morales, A.R. Immunohistochemistry of estrogen and progesterone receptors reconsidered: Experience with 5,993 breast cancers. Am. J. Clin. Pathol. 2005, 123, 21–27. [Google Scholar] [CrossRef]
- Sun, D.; Chen, G.; Dellinger, R.W.; Duncan, K.; Fang, J.L.; Lazarus, P. Characterization of tamoxifen and 4-hydroxytamoxifen glucuronidation by human UGT1A4 variants. Breast Cancer Res. 2006, 8, R50. [Google Scholar] [CrossRef] [PubMed]
- Pratama, M.R.F.P.; Poerwono, H.; Siswandono, S. Design and molecular docking of novel 5-O-Benzoylpinostrobin derivatives as anti-breast cancer. TJPS 2019, 43, 201–212. [Google Scholar]
- Prasetiawati, R.; Zamri, A.; Barliana, M.I.; Muchtaridi, M. In silico predictive for modification of chalcone with pyrazole derivatives as a novel therapeutic compound for targeted breast cancer treatment. J. Appl. Pharm. Sci. 2019, 9, 20–28. [Google Scholar]
- Herfindo, N.; Prasetiawati, R.; Sialagan, D.; Frimayanti, N.; Zamri, A. Synthesis, antiproliferative activity and molecular docking studies of 1,3,5-triaryl pyrazole compound as estrogen α receptor inhibitor targeting MCF-7 cells line. Molekul 2020, 15, 18–25. [Google Scholar] [CrossRef]
- Frimayanti, N.; Yaeghoobi, M.; Namavar, H.; Ikhtiarudin, I.; Afzali, M. In silico studies and biological evaluation of chalcone-based 1,5-benzothiazepines as new potential H1N1 neuraminidase inhibitors. J. Appl. Pharm. Sci. 2020, 10, 86–94. [Google Scholar] [CrossRef]
- Jasril, J.; Ikhtiarudin, I.; Hasti, S.; Reza, A.I.; Frimayanti, N. Microwave-assisted synthesis, in silico studies and in vivo evaluation for antidiabetic activity of new brominated pyrazoline analogs. TJPS 2019, 43, 83–89. [Google Scholar]
- Jasril, J.; Ikhtiarudin, I.; Zamri, A.; Teruna, H.Y.; Frimayanti, N. New fluorinated chalcone and pyrazoline analogs: Synthesis, docking, and molecular dynamic studies as anticancer agents. TJPS 2017, 41, 93–98. [Google Scholar]
- Zamri, A.; Teruna, H.Y.; Wulansari, S.; Herfindo, N.; Frimayanti, N.; Ikhtiarudin, I. 3-(3,4-Dimethoxyphenyl)-5-(2-fluorophenyl)-1-phenyl-4,5-dihydro-1H-pyrazole. Molbank 2019, 2019, M1088. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbons | 1H NMR δ (ppm), J (Hz) | 13C NMR δ (ppm) | 1H-1H COSY | Important 1H-13C HMBC |
|---|---|---|---|---|
| 1 | - | 112.46 | - | - |
| 2 | 10.82 (s, 1H, OH) | 161.67 | - | 112.46 (C1), 161.67(C2), 117.58 (C3) |
| 3 | 6.99 (d, 1H, J = 8.5 Hz) | 117.58 | 4 | |
| 4 | 7.46 (dt, 1H, J1 = 8 Hz, J2 = 1.5 Hz) | 135.67 | 3, 5 | |
| 5 | 6.87 (t, 1H, J = 7.5 Hz) | 119.14 | 4, 6 | |
| 6 | 7.84 (dd, 1H, J1 = 8 Hz, J2 = 1.5 Hz) | 129.82 | 5 | |
| C=O ester | - | 170.15 | - | - |
| 1′ | 4.46 (t, 2H, J = 5.5 Hz) | 64.98 | 2′ | 170.15 (C=O ester) |
| 2′ | 2.01 (m, 2H) | 25.44 | 1′ | |
| 3′ | 2.01 (m, 2H) | 25.90 | 4′ | |
| 4′ | 4.13 (t, 2H, J = 5.5 Hz) | 67.42 | 3′ | 159.13 (C3′) |
| 1′′ | - | 159.13 | - | - |
| 2′′ | 7.55 (s, 1H) | 113.47 | 3′′ | 190.19 (C=O ketone) |
| 3′′ | - | 139.92 | - | - |
| 4′′ | 7.60 (d, 1H, J = 6 Hz) | 121.00 | 5′′ | 190.19 (C=O ketone) |
| 5′′ | 7.41 (t, 1H, J = 8 Hz) | 119.76 | 4′′ 6′′ | 139.92 (C3′′), 159.13 (C1′′) |
| 6′′ | 7.12 (dd, 1H, J1 = 8 Hz, J2 = 2 Hz) | 119.41 | 5′′ 2′′ | |
| C=O ketone | - | 190.19 | - | - |
| Cα | 7.40 (d, 1H, J = 15.5 Hz) | 129.54 | β | 190.19 (C=O ketone), 127.60 (C1′′′), 130.24 (C2′′′) |
| Cβ | 7.80 (d, 1H, J= 15.5 Hz) | 144.72 | α | 190.19 (C=O ketone), 139.92 (C3”); 127.60 (C1′′′) |
| 1′′′ | - | 127.60 | - | - |
| 2′′′/6′′′ | 7.61 (d, 2H, J = 8.5 Hz) | 130.24 | 3′′′/5′′′ | 144.72 (Cβ); 161.70 (C4′′′) |
| 3′′′/5′′′ | 6.95 (d, 2H, J = 8.5 Hz); | 114.42 | 2′′′/6′′′ | 127.60 (C1′′′) |
| 4′′′ | - | 161.70 | - | - |
| OCH3 | 3.87 (s, 3H) | 55.41 | - | 161.70 (C4′′′) |
| Compounds | ∆G (kcal/mol) | Interactions with Amino Acid Residues | ||
|---|---|---|---|---|
| H Bond | van der Walls | Other Hydrophobic Interactions | ||
| Chalcone analogue | −6.32 | Glu353 | Arg394, Leu428, Met388, Ile424, Met421, Gly521, Gly420, Glu419, Thr347 | His524, Leu525, Leu384, Met343, Leu349, Phe404, Leu346, Leu391, Leu387, Ala350 |
| Title Compound | −8.15 | Cys530 | Ile424, Phe404, Val533, Met528, Lys529, Val534, Leu354, Leu536, Trp383, Leu346 | Leu387, Asp351, Thr347, Leu384, Met343, Ala350, Met388, Leu391, Leu428, Leu525, Met421, Leu539 |
| Tamoxifen | −7.00 | - | Arg394, Glu353, Met517, Met388, Ile424, Gly521, Lys520, Gly521, His524, Gly420, Leu354, Met528, Trp383, Thr347, Leu536, Leu428, Phe404 | Leu525, Asp351, Leu384, Met421, Met343, Leu349, Leu346, Leu387, Leu391, Ala350 |
| 4-OHT | −9.02 | Glu353 | Glu419, Ile424, Gly420, His524, Gly521, Met522, Arg394, Phe404, Thr347, Lys529, Trp383, Leu536, Leu354, Leu539 | Asp351, Leu525, Met421, Leu428, Met388, Leu391, Met343, Ala350, Leu384, Leu387, Leu346, Leu349 |
| Amino Acid Residues | |||
|---|---|---|---|
| H Bond | van der Walls | Other Hydrophobic Interactions | |
| Before MD simulation | Cys530 | Ile424, Phe404, Val533, Met528, Lys529, Val534, Leu354, Leu536, Trp383, Leu346 | Leu387, Asp351, Thr347, Leu384, Met343, Ala350, Met388, Leu391, Leu428, Leu525, Met421, Leu539 |
| After MD simulation | Asp351, Thr347 | Met388, Gly521, Leu384, Leu525, Leu536, Ile358, Leu540, Leu539, Met543, Asn532, Gly420 | His524, Pro535, Ile424, Met421, Leu346, Ala350, Leu354,Val534, Lys529, Trp383 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikhtiarudin, I.; Dona, R.; Frimayanti, N.; Utami, R.; Susanti, E.; Mentari, M.; Nurmaida, N.; Agistia, N.; Herfindo, N.; Zamri, A. (E)-4-(3-(3-(4-Methoxyphenyl)acryloyl)phenoxy)butyl 2-Hydroxybenzoate. Molbank 2021, 2021, M1195. https://doi.org/10.3390/M1195
Ikhtiarudin I, Dona R, Frimayanti N, Utami R, Susanti E, Mentari M, Nurmaida N, Agistia N, Herfindo N, Zamri A. (E)-4-(3-(3-(4-Methoxyphenyl)acryloyl)phenoxy)butyl 2-Hydroxybenzoate. Molbank. 2021; 2021(1):M1195. https://doi.org/10.3390/M1195
Chicago/Turabian StyleIkhtiarudin, Ihsan, Rahma Dona, Neni Frimayanti, Rahayu Utami, Emma Susanti, Mentari Mentari, Nurmaida Nurmaida, Nesa Agistia, Noval Herfindo, and Adel Zamri. 2021. "(E)-4-(3-(3-(4-Methoxyphenyl)acryloyl)phenoxy)butyl 2-Hydroxybenzoate" Molbank 2021, no. 1: M1195. https://doi.org/10.3390/M1195
APA StyleIkhtiarudin, I., Dona, R., Frimayanti, N., Utami, R., Susanti, E., Mentari, M., Nurmaida, N., Agistia, N., Herfindo, N., & Zamri, A. (2021). (E)-4-(3-(3-(4-Methoxyphenyl)acryloyl)phenoxy)butyl 2-Hydroxybenzoate. Molbank, 2021(1), M1195. https://doi.org/10.3390/M1195