
Tetramethyl 1,1′-(2-[{4,5-bis(Methoxycarbonyl)-1H-1,2,3-triazol-1-yl}methyl]-2-[(4-methylphenyl)sulfonamido]propane-1,3-diyl)bis(1H-1,2,3-triazole-4,5-dicarboxylate)

 ,
,
Abstract
1. Introduction
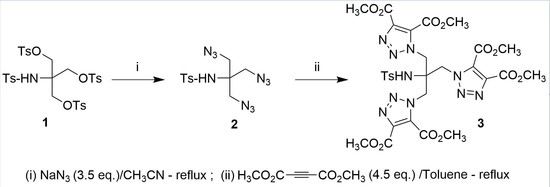
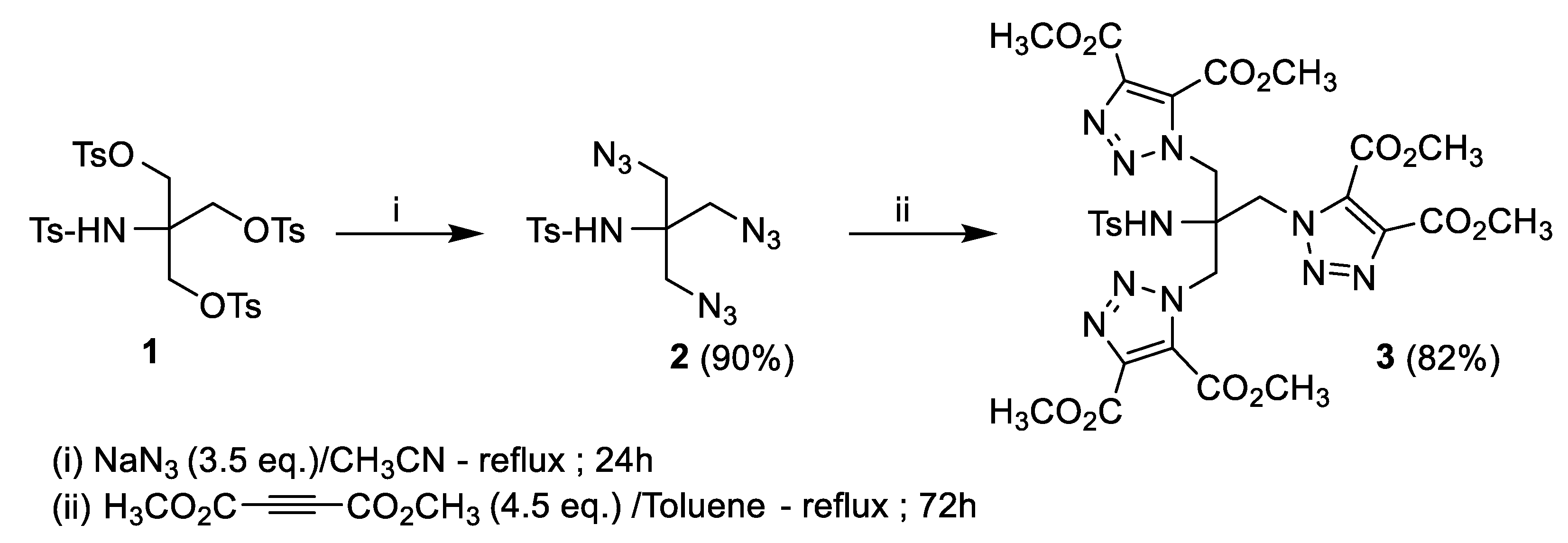
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of N-(1,3-Diazido-2-(azidomethyl)propan-2-yl)-4-methylbenzenesulfonamide (2)
3.2. Synthesis of Tetramethyl 1,1′-(2-[{4,5-bis(Methoxycarbonyl)-1H-1,2,3-triazol-1-yl}methyl]-2-[(4-methylphenyl)sulfonamido]propane-1,3-diyl)bis(1H-1,2,3-triazole-4,5-dicarboxylate) (3)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zarei, S.; Komeili, G.; Bahadorikhalili, S.; Yahya-Meymandi, A.; Karami-Zarandi, M.; Larijani, B.; Biglar, M.; Sadat Ebrahimi, S.E.; Mahdavi, M. Design, synthesis and antibacterial activity evaluation of novel 2-(4-((1-aryl-1H-1,2,3-triazol-4-yl)methoxy)phenyl)2-(2-oxoazetidin-1-yl)acetamide derivatives. J. Heterocycl. Chem. 2020, 57, 4254–4261. [Google Scholar] [CrossRef]
- Singh, G.; Singh, J.; Singh, A.; Singh, J.; Kumar, M.; Gupta, K.; Chhibber, S. Synthesis, characterization and antibacterial studies of schiff based 1,2,3-triazole bridged silatranes. J. Organomet. Chem. 2018, 871, 21–27. [Google Scholar] [CrossRef]
- Rani, A.; Singh, G.; Singh, A.; Maqbool, U.; Kaur, G.; Singh, J. CuAAC-ensembled 1,2,3-triazole-linked isosteres as pharmacophores in drug discovery: Review. RSC Adv. 2020, 10, 5610–5635. [Google Scholar] [CrossRef]
- Kaushik, C.P.; Luxmi, R.; Kumar, M.; Singh, D.; Kumar, K.; Pahwa, A. One-pot facile synthesis, crystal structure and antifungal activity of 1,2,3-triazoles bridged with amine-amide functionalities. Synth. Commun. 2019, 49, 118–128. [Google Scholar] [CrossRef]
- González-Olvera, R.; Espinoza-Vázquez, A.; Negrón-Silva, G.E.; Palomar-Pardavé, M.E.; Romero-Romo, M.A.; Santillan, R. Multicomponent click synthesis of new 1,2,3-triazole derivatives of pyrimidine nucleobases: Promising acidic corrosion inhibitors for steel. Molecules 2013, 18, 15064–15079. [Google Scholar] [CrossRef] [PubMed]
- Curtius, T. Ueber die Einwirkung von salpetriger Säure auf salzsauren Glycocolläther. Ber. der Dtsch. Chem. Ges. 1883, 16, 2230–2231. [Google Scholar] [CrossRef]
- Buchner, E. Einwirkung von diazoessigäther auf die aether ungesättigter säuren. Ber. der Dtsch. Chem. Ges. 1888, 21, 2637–2647. [Google Scholar] [CrossRef]
- 1,3‐Dipolar cycloaddition chemistry. Volumes 1 and 2. Edited by Albert Padwa. John Wiley and Sons. New York, 1984. Volume 1: XIII + 817 pages. Volume 2: XIII + 704 pages. J. Heterocycl. Chem. 1986, 23, 1899. [CrossRef]
- Suga, H. Synthetic Applications of 1,3‐Dipolar Cycloaddition Chemistry. Towards Heterocycles and Natural Products (Series: Chem-istry of Heterocyclic Compounds, Vol. 59). Angew. Chem. Int. Ed. 2003, 42, 4569–4570. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-Dipolar cycloadditions. Past and Future. Angew. Chem. Int. Ed. Engl. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Atmani, A.; El Hallaoui, A.; El Hajji, S.; Roumestant, M.L.; Viallefont, P. From oxazolines to precursors of amino acids. Synth. Commun. 1991, 21, 2383–2390. [Google Scholar] [CrossRef]
- Labriti, B.; El Hallaoui, A.; Elachqar, A.; Alami, A.; El Hajji, S.; Boukallaba, K.; El Bali, B.; Lachkar, M.; Allouchi, H.; Martinez, J.; et al. Synthesis of 2-phenyl-4-methyl-4-((tetrazol-5-yl)methyl) oxazoline. Mor. J. Heterocycl. Chem. 2006, 5, 58–61. [Google Scholar] [CrossRef]
- Aouine, Y.; Faraj, H.; Alami, A.; El Hallaoui, A.; Elachqar, A.; El Hajji, S.; Kerbal, A.; Labriti, B.; Martinez, J.; Rolland, V. Synthesis of new triheterocyclic compounds, precursors of biheterocyclic aminoacids. Mor. J. Heterocycl. Chem. 2008, 7, 44–49. [Google Scholar] [CrossRef]
- Aouine, Y.; Faraj, H.; Alami, A.; El Hallaoui, A.; El Hajji, S.; Labriti, B.; Kerbal, A. Triheterocyclic compounds, oxazolinic precursors of biheterocyclic amino acids, Part II: Phenothiazine derivatives and structural study of regioisomers through 1H-15N 2D NMR HMBC. Mor. J. Heterocycl. Chem. 2014, 13, 39–47. [Google Scholar] [CrossRef]
- Younas, A.; Abdelilah, E.H.; Anouar, A. N,N-Dibenzyl-1-(1-[(4-methyl-2-phenyl-4,5-dihydrooxazol-4-yl)methyl)]-1H-1,2,3-triazol-4-yl)methanamine. Molbank 2014, 2014, M819. [Google Scholar] [CrossRef]
- Achamlale, S.; Alami, A.; Aouine, Y. Structure assignment of N-protected 2-(1H-1,2,3- triazol-1-yl)-glycine derivatives by chemical and spectroscopic methods. Mor. J. Heterocycl. Chem. 2019, 18, 61–69. [Google Scholar] [CrossRef]
- Boukhssas, S.; Aouine, Y.; Faraj, H.; Alami, A.; El Hallaoui, A.; Bekkari, H. Synthesis, characterization, and antibacterial activity of diethyl 1-((4-methyl-2-phenyl-4,5-dihydrooxazol-4-yl)methyl)-1H-1,2,3-triazole-4,5-dicarboxylate. J. Chem. 2017, 2017, 4238360. [Google Scholar] [CrossRef]
- Achamlale, S.; Elachgar, A.; El Hallaoui, A.; Alami, A.; El Hajji, S.; Roumestant, M.L.; Viallefont, P. Synthesis of biheterocyclic α-amino acids. Amino Acids 1999, 17, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Dioukhane, K.; Moussaid, S.; Achamlale, S.; Alami, A.; Kabbour, M.R.; Aouine, Y.; Faraj, H.; Bouksaim, M.; Gaye, M.L. Comparative study of antibacterial activity of some biheterocyclic “triazolic-tetrazolic” α-amino acid derivatives against bacterial strains. Mor. J. Heterocycl. Chem. 2020, 19, 87–91. [Google Scholar] [CrossRef]
- Hajib, S.; Ksakas, A.; Aouine, Y.; Alami, A.; Faraj, H. Synthesis and structural determination of two new tri-heterocyclic regioisomeric compounds, precursors of bi-triazolic α-amino acids, via a comparative study using 1D NMR Spectroscopy. Eur. J. Adv. Chem. Res. 2020, 1, 1–6. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
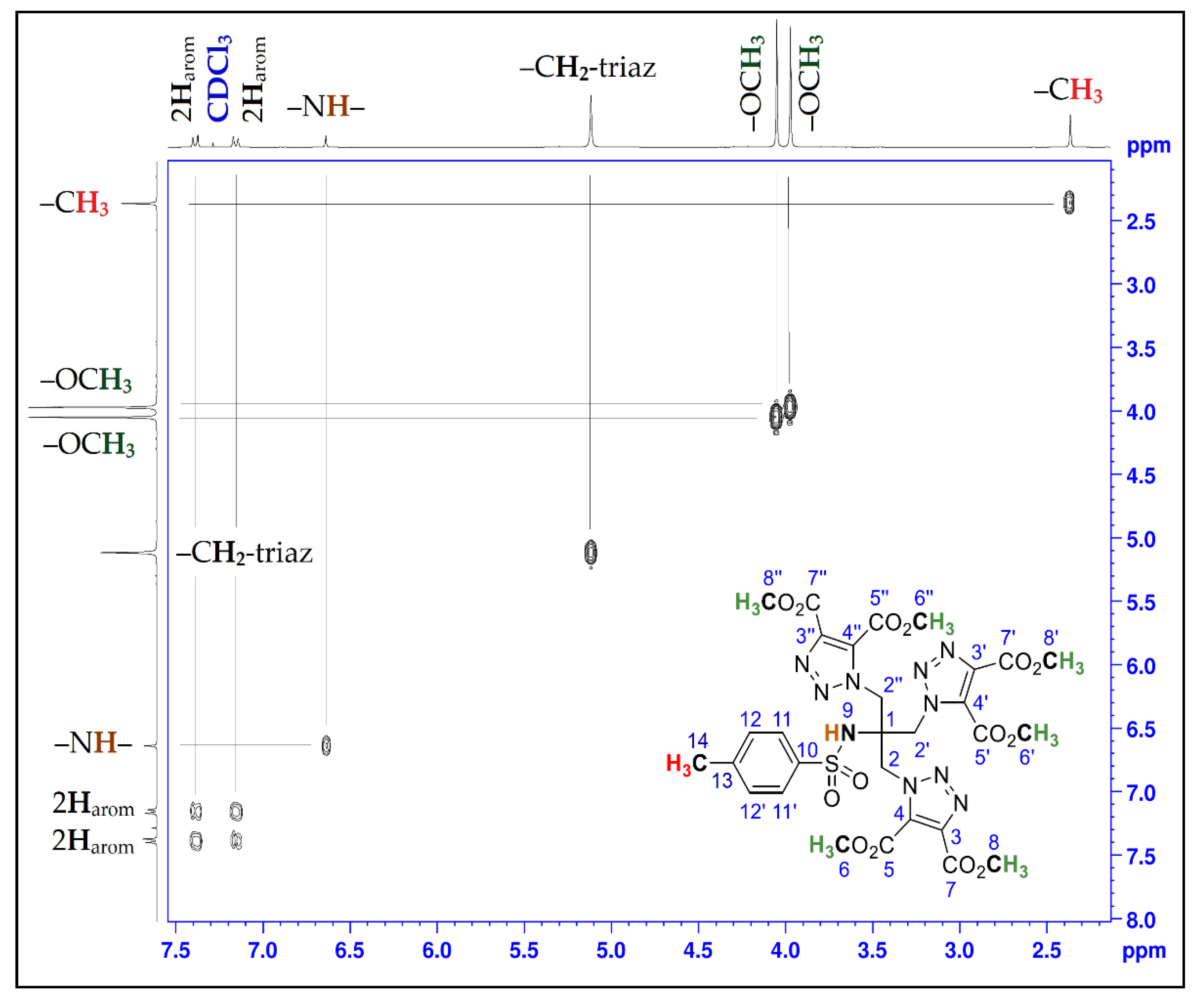
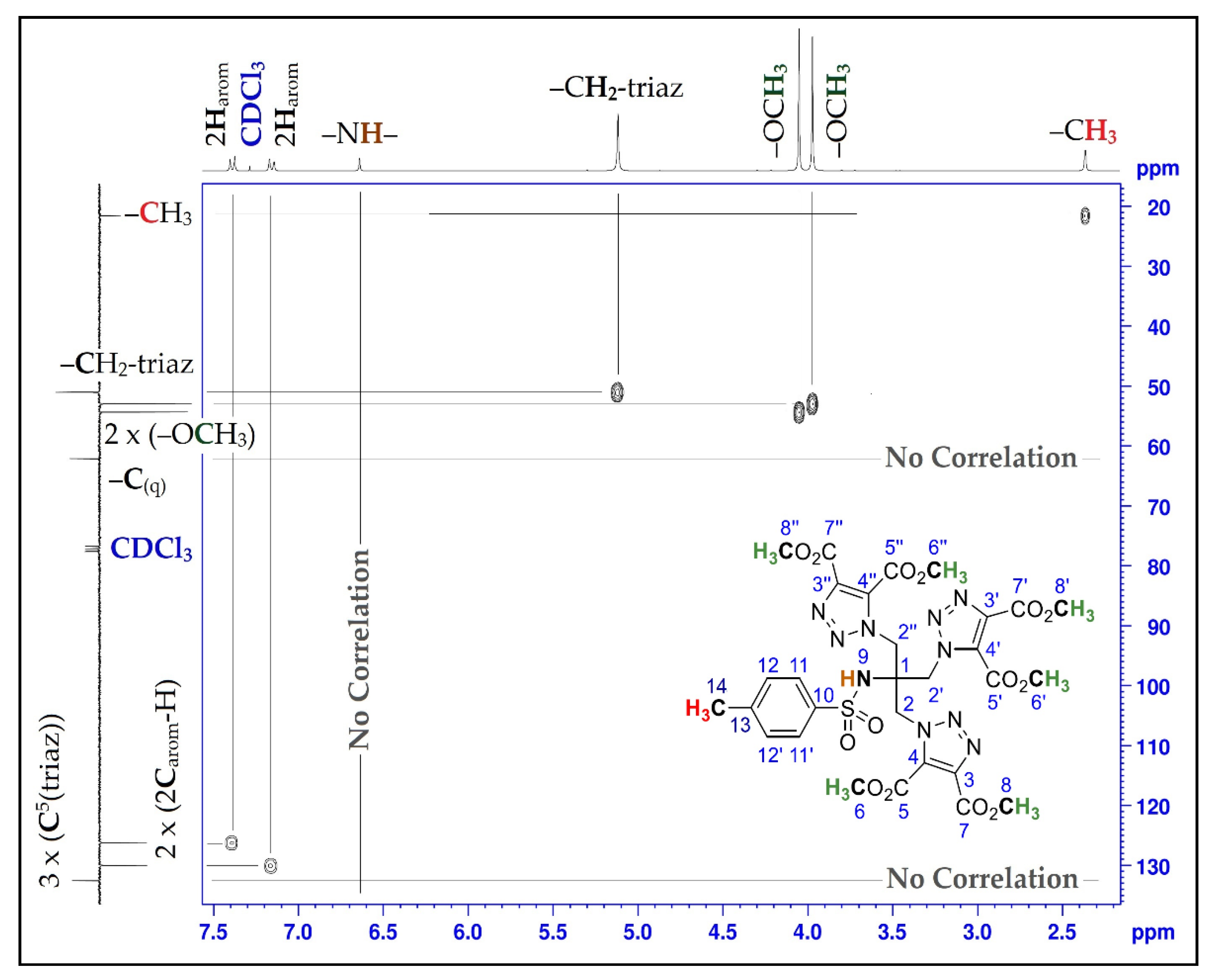
| Position | δH | δC | Correlation H-H | Correlation C-H |
|---|---|---|---|---|
| 1 | - | 62.1 | - | - |
| 2; 2′; 2″ | 5.12 (s) | 51.1 | 2H2-2H2; 2H2′-2H2′; 2H2″-2H2″ | C2-2H2; C2′-2H2′; C2″-2H2″ |
| 3; 3′; 3″ | - | 139.6 | - | - |
| 4; 4′; 4″ | - | 132.5 | - | - |
| 5; 5′; 5″ | - | 159.1 | - | - |
| 6; 6′; 6″ | 4.05 (s) | 52.9 | 3H6-3H6; 3H6′-3H6′; 3H6″-3H6″ | C6-3H6; C6′-3H6′; C6″-3H6″ |
| 7; 7′; 7″ | - | 160.0 | - | - |
| 8; 8′; 8″ | 3.97 (s) | 54.3 | 3H8-3H8; 3H8′-3H8′; 3H8′′-3H8″ | C8-3H8; C8′-3H8′; C8″-3H8″ |
| 9 | 6.64 (s) | - | - | - |
| 10 | - | 144.3 | - | - |
| 11; 11′ | 7.16 (d, J = 8.25) | 126.1 | 1H11-1H11; 1H11′-1H11′ | C11-1H11; C11′-1H11′ |
| 12; 12′ | 7.38 (d, J = 8.34) | 130.0 | 1H12-1H12; 1H12′-1H12′ | C12-1H12; C12′-1H12′ |
| 13 | - | 138.4 | - | - |
| 14 | 2.37 (s) | 21.5 | 3H14-3H14 | C14-3H14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fall, S.A.K.; Hajib, S.; Karai, O.; Aouine, Y.; Boukhssas, S.; Faraj, H.; Alami, A. Tetramethyl 1,1′-(2-[{4,5-bis(Methoxycarbonyl)-1H-1,2,3-triazol-1-yl}methyl]-2-[(4-methylphenyl)sulfonamido]propane-1,3-diyl)bis(1H-1,2,3-triazole-4,5-dicarboxylate). Molbank 2021, 2021, M1186. https://doi.org/10.3390/M1186
Fall SAK, Hajib S, Karai O, Aouine Y, Boukhssas S, Faraj H, Alami A. Tetramethyl 1,1′-(2-[{4,5-bis(Methoxycarbonyl)-1H-1,2,3-triazol-1-yl}methyl]-2-[(4-methylphenyl)sulfonamido]propane-1,3-diyl)bis(1H-1,2,3-triazole-4,5-dicarboxylate). Molbank. 2021; 2021(1):M1186. https://doi.org/10.3390/M1186
Chicago/Turabian StyleFall, Serigne Abdou Khadir, Sara Hajib, Oumaima Karai, Younas Aouine, Salaheddine Boukhssas, Hassane Faraj, and Anouar Alami. 2021. "Tetramethyl 1,1′-(2-[{4,5-bis(Methoxycarbonyl)-1H-1,2,3-triazol-1-yl}methyl]-2-[(4-methylphenyl)sulfonamido]propane-1,3-diyl)bis(1H-1,2,3-triazole-4,5-dicarboxylate)" Molbank 2021, no. 1: M1186. https://doi.org/10.3390/M1186
APA StyleFall, S. A. K., Hajib, S., Karai, O., Aouine, Y., Boukhssas, S., Faraj, H., & Alami, A. (2021). Tetramethyl 1,1′-(2-[{4,5-bis(Methoxycarbonyl)-1H-1,2,3-triazol-1-yl}methyl]-2-[(4-methylphenyl)sulfonamido]propane-1,3-diyl)bis(1H-1,2,3-triazole-4,5-dicarboxylate). Molbank, 2021(1), M1186. https://doi.org/10.3390/M1186