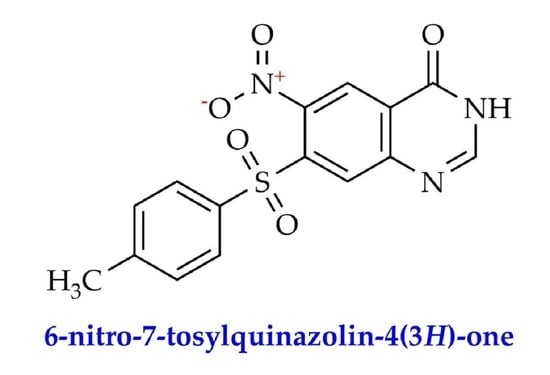
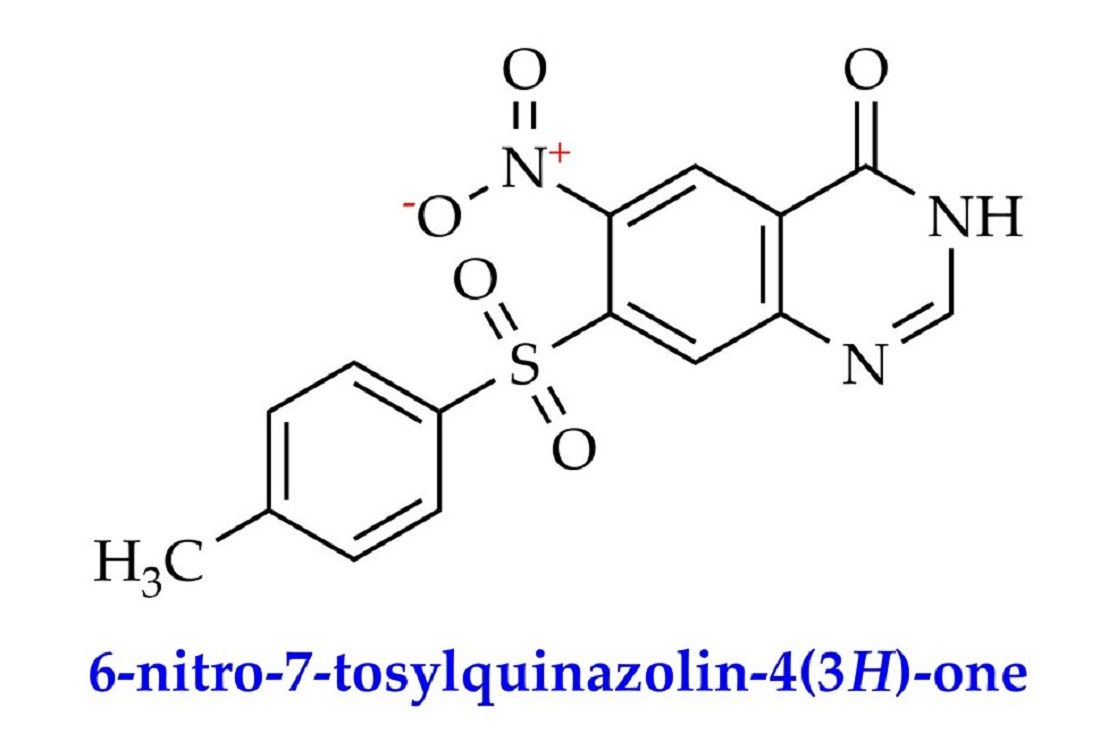
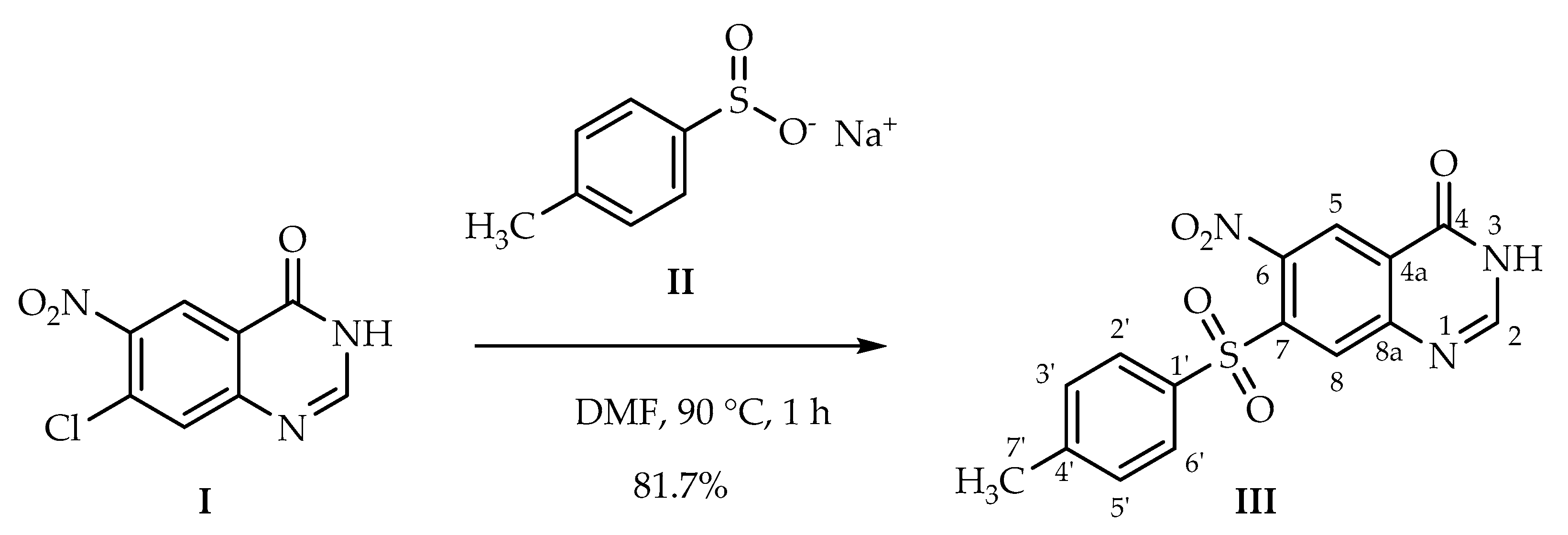
6-Nitro-7-tosylquinazolin-4(3H)-one



Abstract
:
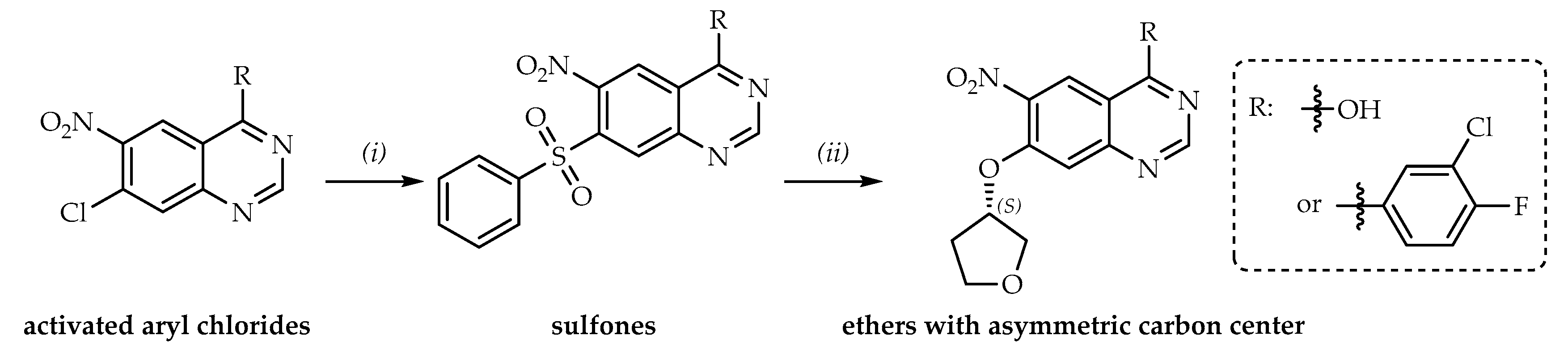
1. Introduction
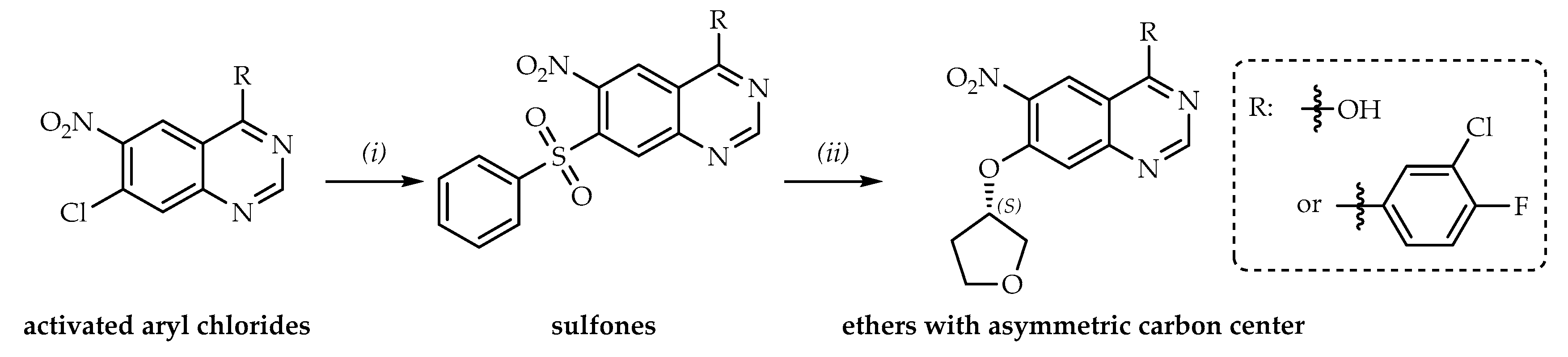
2. Results and Discussion
3. Materials and Methods
3.1. General Information
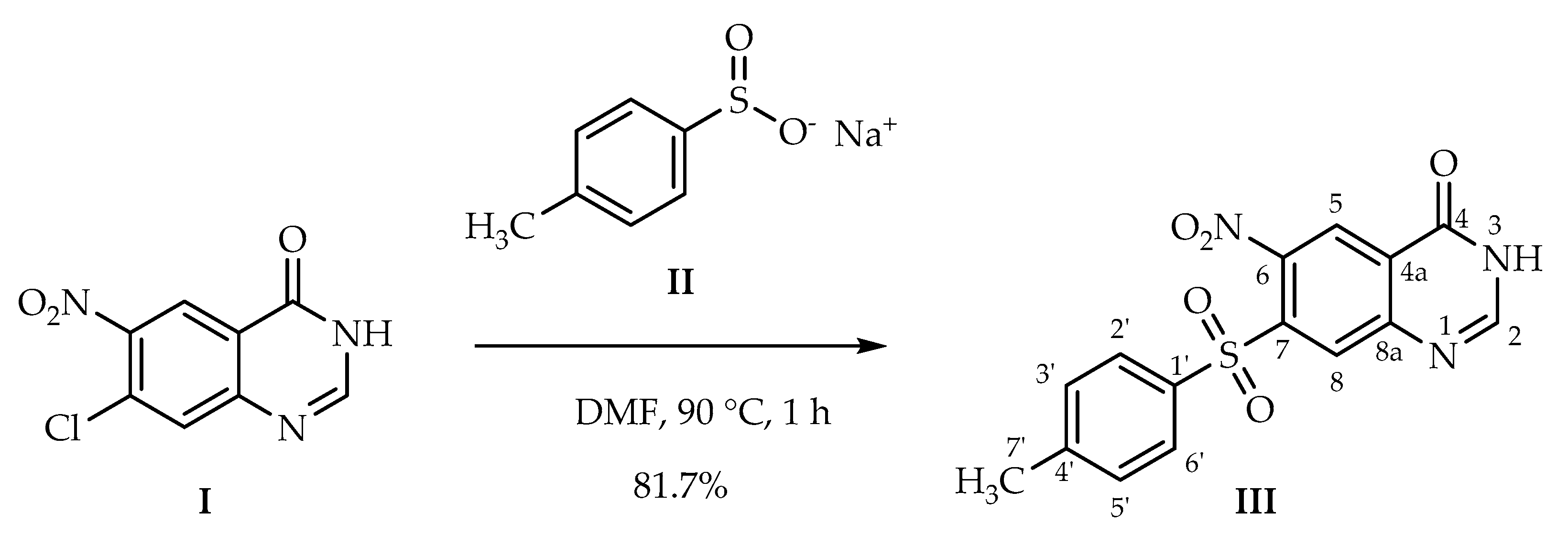
3.2. Synthetic Procedure
6-Nitro-7-tosylquinazolin-4(3H)-one (III)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, N.-W.; Liang, S.; Manolikakes, G. Recent advances in the synthesis of sulfones. Synthesis 2016, 48, 1939–1973. [Google Scholar] [CrossRef] [Green Version]
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur containing scaffolds in drugs: Synthesis and application in medicinal chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef]
- Matavos-Aramyan, S.; Soukhakian, S.; Jazebizadeh, M.H. Selected methods for the synthesis of sulfoxides and sulfones with emphasis on oxidative protocols. Phosphorus Sulfur Silicon Relat. Elem. 2020, 195, 181–193. [Google Scholar] [CrossRef]
- The DrugBank Database. Available online: https://www.drugbank.ca (accessed on 18 May 2020).
- Richards-Taylor, C.S.; Blakemore, D.C.; Willis, M.C. One-pot three-component sulfone synthesis exploiting palladium-catalysed aryl halide aminosulfonylation. Chem. Sci. 2014, 5, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Trost, B.M.; Kalnmals, C.A. Sulfones as chemical chameleons: Versatile synthetic equivalents of small-molecule synthons. Chem. Eur. J. 2019, 25, 11193–11213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noser, A.A.; El-Naggar, M.; Donia, T.; Abdelmonsef, A.H. Synthesis, in silico and in vitro assessment of new quinazolinones as anticancer agents via potential AKT inhibition. Molecules 2020, 25, 4780. [Google Scholar] [CrossRef] [PubMed]
- El-Azab, A.S.; Abdel-Aziz, A.A.; AlSaif, N.A.; Alkahtani, H.M.; Alanazi, M.M.; Obaidullah, A.J.; Eskandrani, R.O.; Alharbi, A. Antitumor activity, multitarget mechanisms, and molecular docking studies of quinazoline derivatives based on a benzenesulfonamide scaffold: Cell cycle analysis. Bioorg. Chem. 2020, 104, 104345. [Google Scholar] [CrossRef] [PubMed]
- Sancineto, L.; Iraci, N.; Massari, S.; Attanasio, V.; Corazza, G.; Barreca, M.L.; Sabatini, S.; Manfroni, G.; Avanzi, N.R.; Cecchetti, V.; et al. Computer--aided design, synthesis and validation of 2--phenylquinazolinone fragments as CDK9 inhibitors with anti--HIV--1 Tat--mediated transcription activity. ChemMedChem 2013, 8, 1941–1953. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Cheng, C.; Xu, G.B.; Tang, L.; Chua, K.L.; Yang, Y.Y. Synthesis and antibiofilm evaluation of 3-hydroxy-2,3-dihydroquinazolin-4(1H)-one derivatives against opportunistic pathogen Acinetobacter Baumannii. Bioorg. Med. Chem. 2020, 28, 115606. [Google Scholar] [CrossRef]
- Liu, G.; Liu, C.P.; Ji, C.N.; Sun, L. Synthesis and antifungal activities of N-3-substituted quinazolin-4-one catalyzed by 3-methylimidazole ionic liquids. Asian J. Chem. 2013, 25, 9853–9856. [Google Scholar] [CrossRef]
- Noureldin, N.A.; Kothayer, H.; Lashine, E.M.; Baraka, M.M.; Huang, Y.; Li, B.; Ji, Q. Design, synthesis and biological evaluation of novel quinazoline-2,4-diones conjugated with different amino acids as potential chitin synthase inhibitors. Eur. J. Med. Chem. 2018, 152, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Aziz, J.; Messaoudi, S.; Alami, M.; Hamze, A. Sulfinate derivatives: Dual and versatile partners in organic synthesis. Org. Biomol. Chem. 2014, 12, 9743–9759. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D.; Klose, I.; Oost, R.; Neuhaus, J.; Maulide, N. Bond-forming and -breaking reactions at sulfur(IV): Sulfoxides, sulfonium salts, sulfur ylides, and sulfinate salts. Chem. Rev. 2019, 119, 8701–8780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tranquilino, A.; Andrade, S.R.C.P.; da Silva, A.P.M.; Menezes, P.H.; Oliveira, R.A. Non-expensive, open-flask and selective catalytic systems for the synthesis of sulfinate esters and thiosulfonates. Tetrahedron Lett. 2017, 58, 1265–1268. [Google Scholar] [CrossRef]
- Li, Y.; Fan, Y. Recent advances in C–S bond construction to synthesize sulfone. Synth. Commun. 2019, 49, 3227–3264. [Google Scholar] [CrossRef]
- Schroeder, J.; Dziewas, G.; Fachinger, T.; Jaeger, B.; Reichel, C.; Renner, S. Process for Preparing Aminocrotonylamino-Substituted Quinazoline Derivatives. U.S. Patent US8188274B2, 29 May 2012. [Google Scholar]
- Slobbe, P.; Poot, A.J.; Van Dongen, A.A.M.S.; Niessen, H.; Windhorst, A.D. Radiolabeled Quinazoline Derivatives. U.S. Patent US20150368230A1, 24 December 2015. [Google Scholar]
- Baskin, J.M.; Wang, Z. An efficient copper catalyst for the formation of sulfones from sulfinic acid salts and aryl iodides. Org. Lett. 2002, 4, 4423–4425. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liang, J.; Chen, Z.; Chen, J.; Yan, M.; Zhang, X. A convenient synthesis of sulfones via light promoted coupling of sodium sulfinates and aryl halides. Adv. Synth. Catal. 2019, 361, 956–960. [Google Scholar] [CrossRef]
- Nguyen, V.D.; Nguyen, V.T.; Haug, G.C.; Dang, H.T.; Arman, H.D.; Ermler, W.C.; Larionov, O.V. Rapid and chemodivergent synthesis of N--heterocyclic sulfones and sulfides: Mechanistic and computational details of the persulfate-initiated catalysis. ACS Catal. 2019, 9, 4015–4024. [Google Scholar] [CrossRef]
- Pretsch, E.; Bühlmann, P.; Badertscher, M. Structure Determination of Organic Compounds, 4th ed.; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Pogaku, N.; Krishna, P.R.; Prapurna, Y.L. Substrate- and temperature-controlled divergence in reactions of alcohols with TosMIC catalyzed by BF3 · Et2O: Facile access to sulfinates and sulfones. Synth. Commun. 2017, 47, 1239–1249. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Tran, T.H.; Dao, N.S.H.; Nguyen, V.G.; Nguyen, D.L.; Trinh, N.T.; Nguyen, V.H. Synthesis and spectral characterization of 4,7-dichloro-6-nitroquinazoline. Molbank 2020, 2020, M1134. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Proton | H-2 | H-5 | H-8 | H-2′ | H-3′ | H-5′ | H-6′ | H-7′ | H-O/H-N | |
|---|---|---|---|---|---|---|---|---|---|---|
| Carbon | ppm | 8.39 | 8.59 | 8.47 | 7.92 | 7.49 | 7.49 | 7.92 | 2.41 | 12.96/3.35 * |
| C-2 | 149.9 | x | γ | |||||||
| C-4 | 159.1 | β | β | |||||||
| C-4a | 126.4 | β | ||||||||
| C-5 | 124.0 | x | γ | |||||||
| C-6 | 144.1 | β | ||||||||
| C-7 | 138.1 | β | ||||||||
| C-8 | 131.2 | γ | γ | |||||||
| C-8a | 151.0 | β | ||||||||
| C-1′ | 136.4 | β | β | |||||||
| C-2′ | 128.1 | x | γ | β | γ | |||||
| C-3′ | 130.1 | x | β | β | ||||||
| C-4′ | 145.5 | β | β | α | ||||||
| C-5′ | 130.1 | β | x | β | ||||||
| C-6′ | 128.1 | β | γ | x | γ | |||||
| C-7′ | 21.1 | β | β | x |
| Peak n | 2θ (°) | d (Å) | I/Imax (%) | Peak n | 2θ (°) | d (Å) | I/Imax (%) |
|---|---|---|---|---|---|---|---|
| 1 | 9.10 | 9.7182 | 2.62 | 33 | 27.50 | 3.2435 | 32.51 |
| 2 | 10.14 | 8.7237 | 15.07 | 34 | 28.00 | 3.1867 | 4.07 |
| 3 | 13.44 | 6.5882 | 12.25 | 35 | 28.38 | 3.1449 | 2.89 |
| 4 | 14.82 | 5.9777 | 32.83 | 36 | 28.62 | 3.1191 | 3.10 |
| 5 | 14.98 | 5.9142 | 21.45 | 37 | 28.84 | 3.0958 | 2.89 |
| 6 | 15.72 | 5.6374 | 2.90 | 38 | 29.44 | 3.0340 | 5.20 |
| 7 | 16.98 | 5.2218 | 3.27 | 39 | 29.58 | 3.0200 | 4.90 |
| 8 | 17.64 | 5.0279 | 18.57 | 40 | 29.98 | 2.9806 | 15.55 |
| 9 | 18.58 | 4.7756 | 2.63 | 41 | 30.36 | 2.9442 | 24.54 |
| 10 | 18.94 | 4.6857 | 17.02 | 42 | 30.64 | 2.9179 | 12.06 |
| 11 | 19.28 | 4.6038 | 2.78 | 43 | 31.68 | 2.8244 | 6.73 |
| 12 | 19.56 | 4.5385 | 3.03 | 44 | 32.32 | 2.7700 | 3.47 |
| 13 | 20.08 | 4.4221 | 5.31 | 45 | 33.04 | 2.7112 | 5.59 |
| 14 | 20.46 | 4.3409 | 7.16 | 46 | 33.58 | 2.6689 | 3.12 |
| 15 | 20.94 | 4.2424 | 12.21 | 47 | 34.72 | 2.5838 | 6.32 |
| 16 | 21.22 | 4.1871 | 4.31 | 48 | 35.26 | 2.5454 | 16.83 |
| 17 | 21.44 | 4.1446 | 5.36 | 49 | 35.82 | 2.5069 | 4.50 |
| 18 | 21.92 | 4.0549 | 32.29 | 50 | 36.48 | 2.4631 | 12.13 |
| 19 | 22.22 | 4.0009 | 7.79 | 51 | 37.32 | 2.4095 | 4.11 |
| 20 | 22.44 | 3.9621 | 6.36 | 52 | 37.70 | 2.3861 | 3.54 |
| 21 | 22.80 | 3.9004 | 56.53 | 53 | 38.24 | 2.3537 | 3.03 |
| 22 | 23.06 | 3.8570 | 15.41 | 54 | 38.44 | 2.3419 | 4.59 |
| 23 | 23.60 | 3.7699 | 11.32 | 55 | 38.68 | 2.3279 | 3.24 |
| 24 | 23.92 | 3.7202 | 20.49 | 56 | 39.10 | 2.3039 | 3.73 |
| 25 | 24.22 | 3.6748 | 100.00 | 57 | 39.86 | 2.2617 | 8.51 |
| 26 | 24.56 | 3.6247 | 7.13 | 58 | 40.68 | 2.2179 | 5.07 |
| 27 | 25.02 | 3.5591 | 4.07 | 59 | 44.48 | 2.0369 | 7.64 |
| 28 | 25.42 | 3.5040 | 5.61 | 60 | 45.32 | 2.0011 | 4.04 |
| 29 | 25.62 | 3.4771 | 4.57 | 61 | 45.60 | 1.9894 | 3.44 |
| 30 | 25.88 | 3.4428 | 4.43 | 62 | 46.56 | 1.9506 | 3.19 |
| 31 | 26.50 | 3.3636 | 21.83 | 63 | 48.48 | 1.8778 | 3.61 |
| 32 | 27.14 | 3.2857 | 5.69 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, T.N.; Tran, T.P.T.; Vu, T.H.M.; Nguyen, H.B.; Dao, N.S.H.; Nguyen, V.G.; Nguyen, D.L.; Trinh, N.T.; Nguyen, V.H. 6-Nitro-7-tosylquinazolin-4(3H)-one. Molbank 2020, 2020, M1168. https://doi.org/10.3390/M1168
Nguyen TN, Tran TPT, Vu THM, Nguyen HB, Dao NSH, Nguyen VG, Nguyen DL, Trinh NT, Nguyen VH. 6-Nitro-7-tosylquinazolin-4(3H)-one. Molbank. 2020; 2020(4):M1168. https://doi.org/10.3390/M1168
Chicago/Turabian StyleNguyen, Thi Ngoc, Thi Phuong Thuy Tran, Thi Hoang Mai Vu, Hoa Binh Nguyen, Nguyet Suong Huyen Dao, Van Giang Nguyen, Dinh Luyen Nguyen, Nguyen Trieu Trinh, and Van Hai Nguyen. 2020. "6-Nitro-7-tosylquinazolin-4(3H)-one" Molbank 2020, no. 4: M1168. https://doi.org/10.3390/M1168
APA StyleNguyen, T. N., Tran, T. P. T., Vu, T. H. M., Nguyen, H. B., Dao, N. S. H., Nguyen, V. G., Nguyen, D. L., Trinh, N. T., & Nguyen, V. H. (2020). 6-Nitro-7-tosylquinazolin-4(3H)-one. Molbank, 2020(4), M1168. https://doi.org/10.3390/M1168