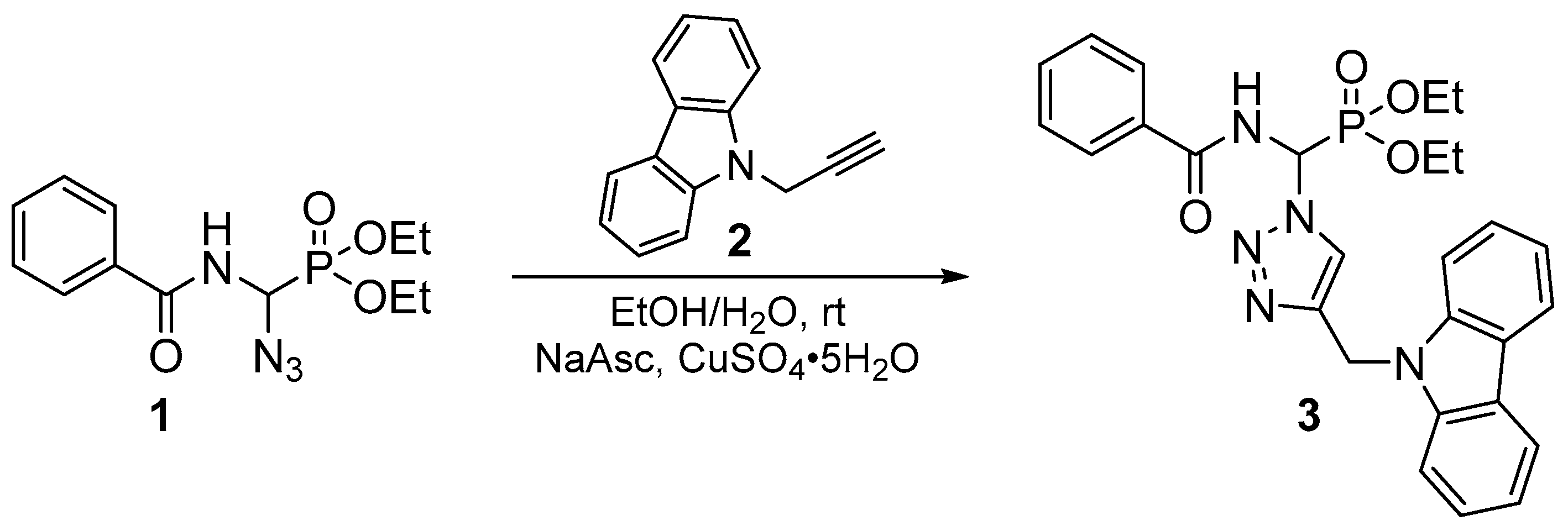
Diethyl [(4-{(9H-carbazol-9-yl)methyl}-1H-1,2,3-triazol-1-yl)(benzamido)methyl]phosphonate


 ,
,  ,
,
Abstract
1. Introduction
2. Results and Discussion
- ◾
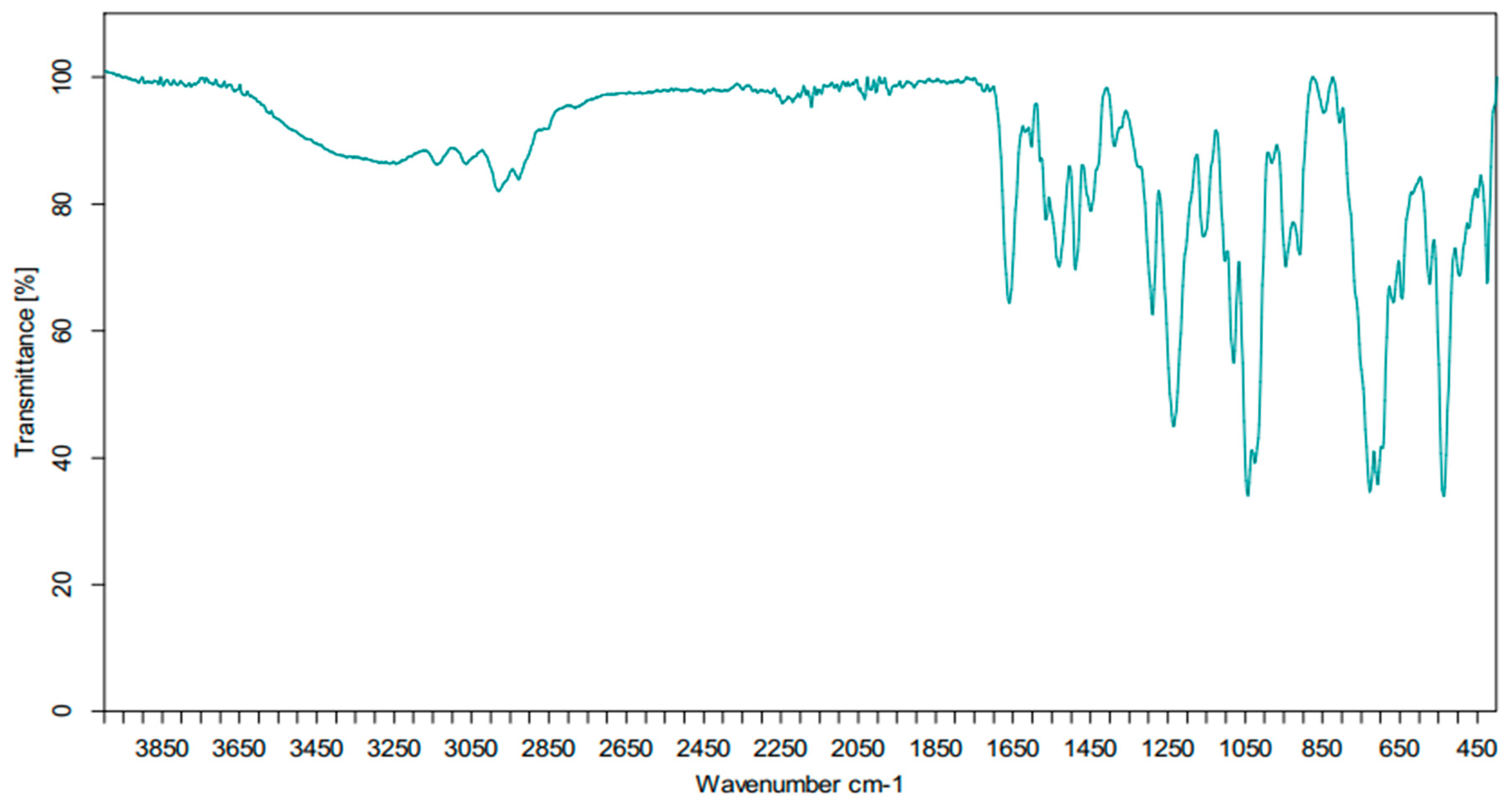
- Two bands in the range of 3050–3100 cm−1, corresponding to the elongation vibrations of ν(Csp2-H).
- ◾
- Two bands in the range of 2900–2950 cm−1, corresponding to the elongation vibrations of ν(Csp3-H).
- ◾
- One intense band around 1650 cm−1, attributable to the C=O function.
- ◾
- A strong band at 1250 cm−1, corresponding to the valence vibrations of the ν(P=O) function.
- ◾
- A medium band at 1050 cm−1, associated with the valence vibrations of the ν(P–O–C) link.
- ◾
- The observation of the bands around 850 and 900 corresponds to the deformation vibrations of ν(N–H).
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Elsherbiny, D.A.; Abdelgawad, A.M.; El-Naggar, M.E.; El-Sherbiny, R.A.; El-Rafie, M.H.; El-Sayed, I.E.-T. Synthesis, antimicrobial activity, and sustainable release of novel α-aminophosphonate derivatives loaded Carrageenan Cryogel. Int. J. Biol. Macromol. 2020, 163, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable potential of the α-aminophosphonate/phosphinate structural motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef] [PubMed]
- Boughaba, S.; Aouf, Z.; Bechiri, O.; Mathe-Allainmat, M.; Lebreton, J.; Aouf, N.-E. H6P2W18O 62·14H2O as an efficient catalyst for the green synthesis of α-aminophosphonates from α-amino acids. Phosphorus Sulfur Silicon Relat. Elem. 2020, 1–8. [Google Scholar] [CrossRef]
- Chiminazzo, A.; Borsato, G.; Favero, A.; Fabbro, C.; McKenna, C.E.; Dalle Carbonare, L.G.; Valenti, M.T.; Fabris, F.; Scarso, A. Diketopyrrolopyrrole bis-phosphonate conjugate: A new fluorescent probe for in vitro bone imaging. Chem. Eur. J. 2019, 25, 3617–3626. [Google Scholar] [CrossRef] [PubMed]
- Rajkoomar, N.; Murugesan, A.; Prabu, S.; Gengan, R.M. Synthesis of methyl piperazinyl-quinolinyl α-aminophosphonates derivatives under microwave irradiation with pd–srtio3 catalyst and their antibacterial and antioxidant activities. Phosphorus Sulfur Silicon Relat. Elem. 2020, 195, 1031–1038. [Google Scholar] [CrossRef]
- Nayab, R.S.; Maddila, S.; Krishna, M.P.; Salam, J.T.; Thaslim, B.S.; Chintha, V.; Wudayagiri, R.; Nagam, V.; Tartte, V.; Chinnam, S. In silico molecular docking and in vitro antioxidant activity studies of novel α-aminophosphonates bearing 6-amino-1,3-dimethyl uracil. J. Recept. Signal Transduct. 2020, 40, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Awad, M.K.; Abdel-Aal, M.F.; Atlam, F.M.; Hekal, H.A. Molecular docking, molecular modeling, vibrational and biological studies of some new heterocyclic α-aminophosphonates. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 206, 78–88. [Google Scholar] [CrossRef]
- Jabli, D.; Dridi, K.; Efrit, M.L. Activité Biologique, réactivité et étude conformationnelle par RMN (1H, 13C, 31P) et DFT des hydrazines phosphonylées vis-à-vis des isothiocyanates. Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 103–108. [Google Scholar] [CrossRef]
- Chafai, N.; Chafaa, S.; Benbouguerra, K.; Daoud, D.; Hellal, A.; Mehri, M. Synthesis, characterization and the inhibition activity of a new α-aminophosphonic derivative on the corrosion of XC48 carbon steel in 0.5M H2SO4: Experimental and theoretical studies. J. Taiwan Inst. Chem. Eng. 2017, 70, 331–344. [Google Scholar] [CrossRef]
- Benbouguerra, K.; Chafaa, S.; Chafai, N.; Mehri, M.; Moumeni, O.; Hellal, A. Synthesis, spectroscopic characterization and a comparative study of the corrosion inhibitive efficiency of an α-aminophosphonate and schiff base derivatives: Experimental and theoretical investigations. J. Mol. Struct. 2018, 1157, 165–176. [Google Scholar] [CrossRef]
- Kaur, G.; Shamim, M.; Bhardwaj, V.; Gupta, V.K.; Banerjee, B. Mandelic acid catalyzed one-pot three-component synthesis of α-aminonitriles and α-aminophosphonates under solvent-free conditions at room temperature. Synth. Commun. 2020, 50, 1545–1560. [Google Scholar] [CrossRef]
- Maestro, A.; Marigorta, E.M.; Palacios, F.; Vicario, J. α-Iminophosphonates: Useful intermediates for enantioselective synthesis of α-aminophosphonates. Asian J. Org. Chem. 2020, 9, 538–548. [Google Scholar] [CrossRef]
- Tripolszky, A.; Tóth, E.; Szabó, P.T.; Hackler, L.; Kari, B.; Puskás, L.G.; Bálint, E. Synthesis and in vitro cytotoxicity and antibacterial activity of novel 1,2,3-triazol-5-yl-phosphonates. Molecules 2020, 25, 2643. [Google Scholar] [CrossRef] [PubMed]
- Elachqar, A.; El Hallaoui, A.; Roumestant, M.L.; Viallefont, P. Synthesis of heterocyclic α-aminophosphonic acids. Synth. Commun. 1994, 24, 1279–1286. [Google Scholar] [CrossRef]
- Aouine, Y.; Faraj, H.; Alami, A.; El Hallaoui, A.; Elachqar, A.; El Hajji, S.; Kerbal, A.; Labriti, B.; Martinez, J.; Rolland, V. Synthesis of new triheterocyclic compounds, precursors of biheterocyclic amino acids. J. Mar. Chim. Heterocycl. 2008, 7, 44–49. [Google Scholar]
- Tripolszky, A.; Németh, K.; Szabó, P.T.; Bálint, E. Synthesis of (1,2,3-triazol-4-yl)methyl phosphinates and (1,2,3-triazol-4-yl)methyl phosphates by copper-catalyzed azide-alkyne cycloaddition. Molecules 2019, 24, 2085. [Google Scholar] [CrossRef]
- Song, W.; Zheng, N.; Li, M.; Ullah, K.; Zheng, Y. Rhodium(I)-catalyzed azide-alkyne cycloaddition (RhAAC) of internal alkynylphosphonates with high regioselectivities under mild conditions. Adv. Synth. Catal. 2018, 360, 2429–2434. [Google Scholar] [CrossRef]
- Huisgen, R. Chimie de la Cycloaddition 1,3-Dipolaire; Wiley: New York, NY, USA, 1984; p. 1. [Google Scholar]
- Bentama, A.; El Hadrami, E.M.; El Hallaoui, A.; Elachqar, A.; Lavergne, J.-P.; Roumestant, M.-L.; Viallefont, P.H. Synthesis of new α-heterocyclic α-amino esters. Amino Acids 2003, 24, 423–426. [Google Scholar] [CrossRef]
- Achamlale, S.; Elachqar, A.; El Hallaoui, A.; El Hajji, S.; Alami, A.; Roumestant, M.L.; Viallefont, Ph. Synthesis of biheterocyclic α-aminophosphonic acid derivatives. Phosphorus Sulfur Silicon 1998, 140, 103–111. [Google Scholar] [CrossRef]
- Boukallaba, K.; Elachqar, A.; El Hallaoui, A.; Alami, A.; El Hajji, S.; Labriti, B.; Rolland, V. Synthesis of new α-heterocyclic α-aminophosphonates. Phosphorus Sulfur Silicon Relat. Elem. 2006, 181, 819–823. [Google Scholar] [CrossRef]
- Boukallaba, K.; Elachqar, A.; El Hallaoui, A.; Alami, A.; El Hajji, S.; Labriti, B.; Rolland, V. Synthesis of α-heterocyclic α-aminophosphonates, part II: Morpholine, piperidine, pyrrolidine, tetrahydrofurylmethylamine, N-benzyl-N-methylamine, and aniline derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2007, 182, 1045–1052. [Google Scholar] [CrossRef]
- Achamlale, S.; Alami, A.; Aouine, Y. Structure assignment of N-protected 2-(1H-1,2,3-triazol-1-yl)-glycine derivatives by chemical and spectroscopic methods. J. Mar. Chim. Heterocycl. 2019, 18, 61–69. [Google Scholar]
- Vorobyeva, D.V.; Karimova, N.M.; Vasilyeva, T.P.; Osipov, S.N.; Shchetnikov, G.T.; Odinets, I.L.; Röschenthaler, G.-V. Synthesis of functionalized α-CF3-α-aminophosphonates via Cu(I)-catalyzed 1,3-dipolar cycloaddition. J. Fluor. Chem. 2010, 131, 378–383. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
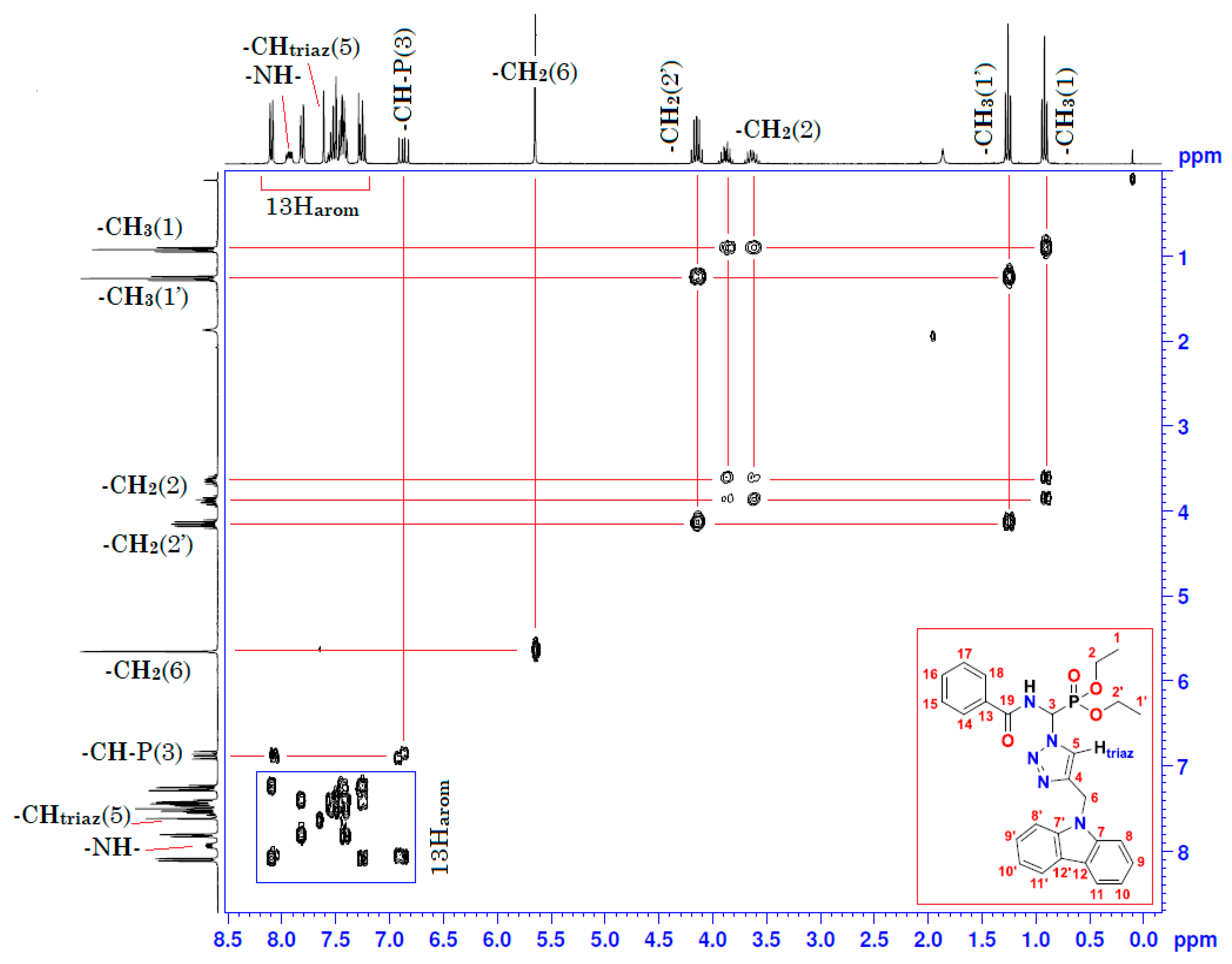
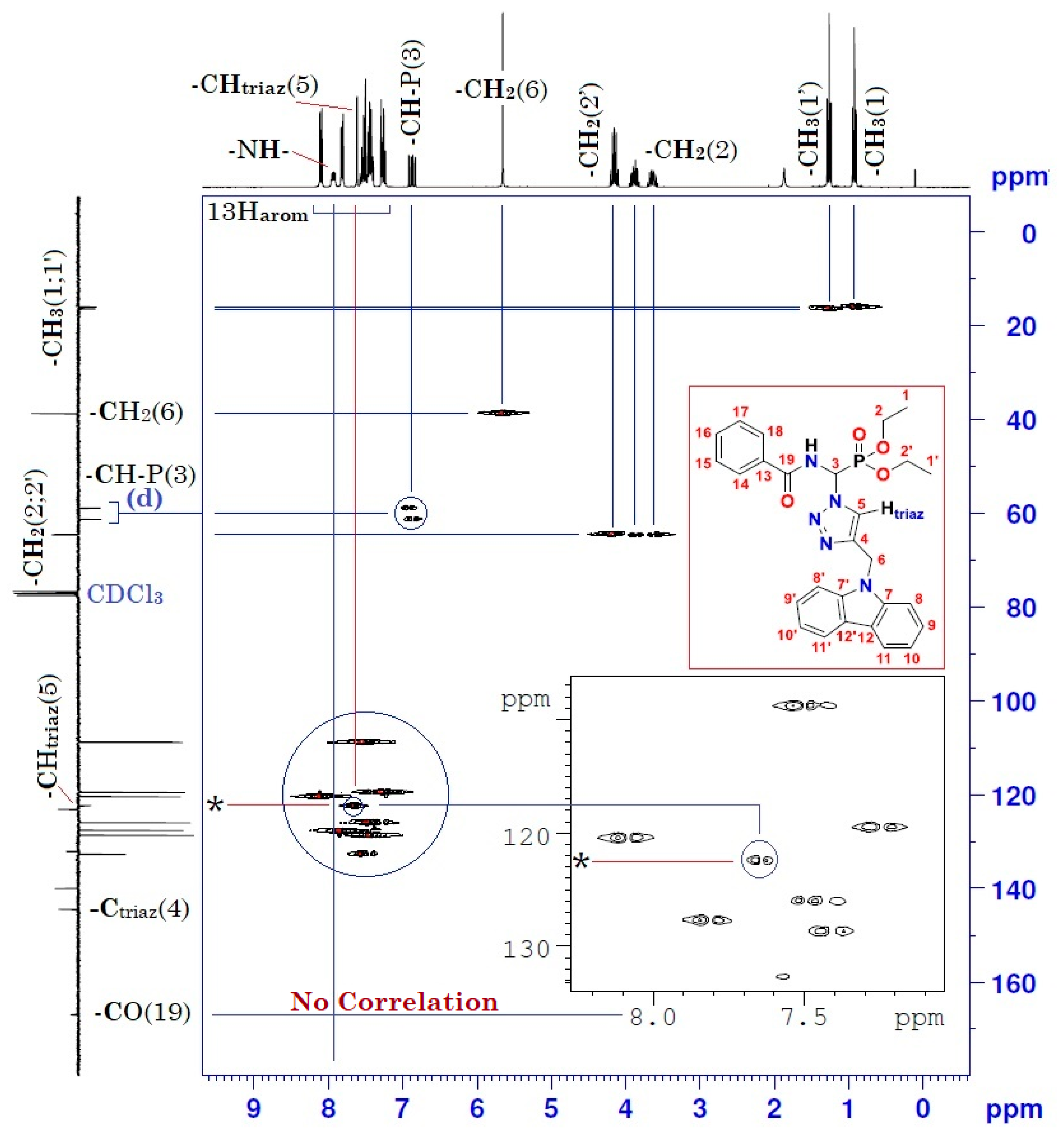
| Position | δH | δC | Correlation H–H | Correlation C–H |
|---|---|---|---|---|
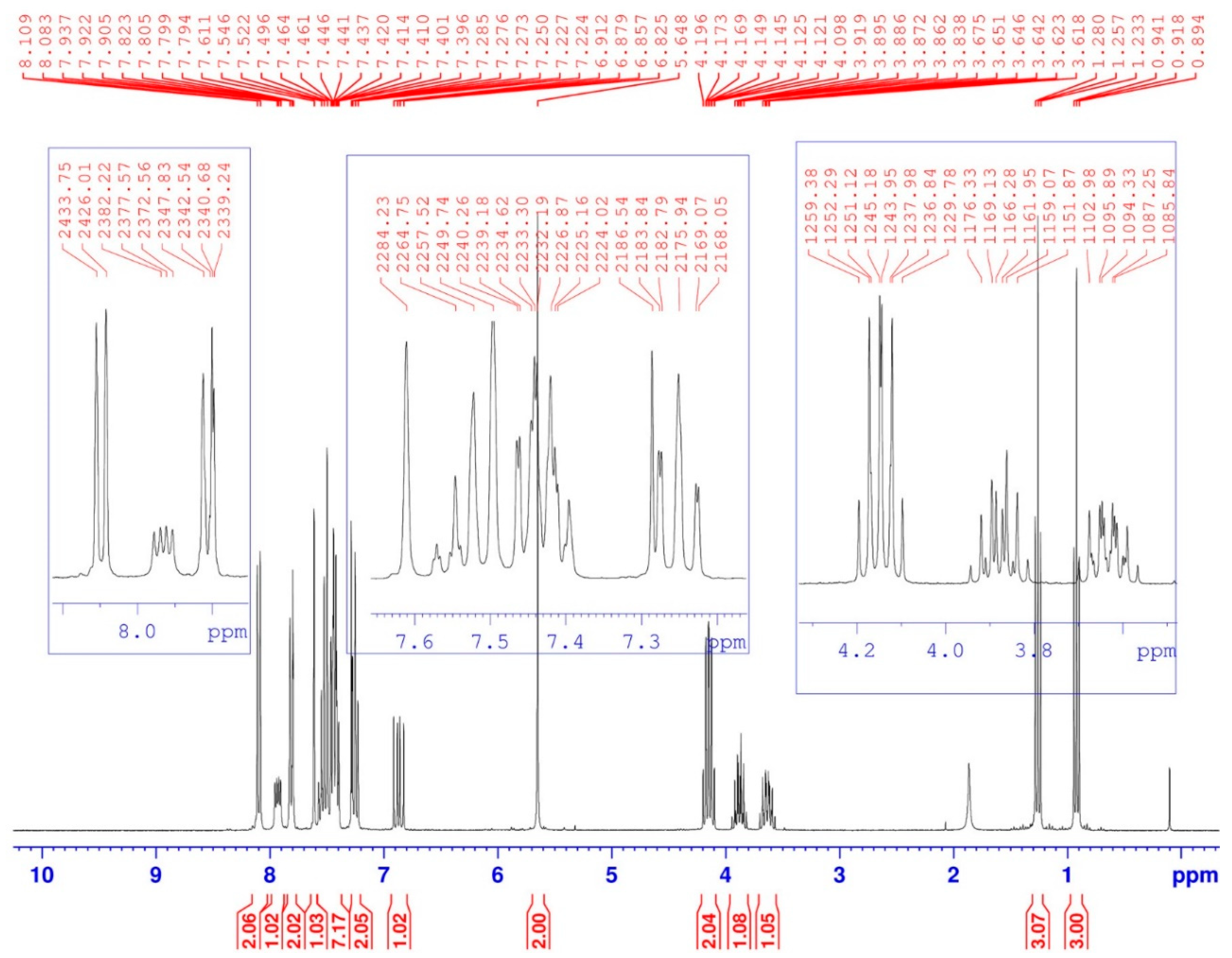
| 1 | 0.92 (t, 3J = 7) | 15.89–15.97 (d, 3JC-P = 5.7) | 3H1-3H1; 3H1-2H2 | C1-3H1 |
| 1′ | 1.26 (t, 3J = 7) | 16.27–16.35 (d, 3JC-P = 6.8) | 3H1′-3H1′; 3H1′-2H2′ | C1′-3H1′ |
| 2 | 3.62–3.67 (m) 3.84–3.92 (m) | 64.49–64.64 (d, 2JC-P = 11.3) | 2H2-2H2; 2H2-3H1 | C2-2H2 |
| 2′ | 4.10–4.20 (m) | 64.40–64.55 (d, 2JC-P = 11.3) | 2H2′-2H2′; 2H2′-3H1′ | C2′-2H2′ |
| 3 | 6.82–6.91 (dd, 3JH-H = 10; 2JH-P = 15.4) | 58.87–61.27 (d, 1JC-P = 181.1) | 1H3-1H3 1H3-1Hamide | C3-1H3 |
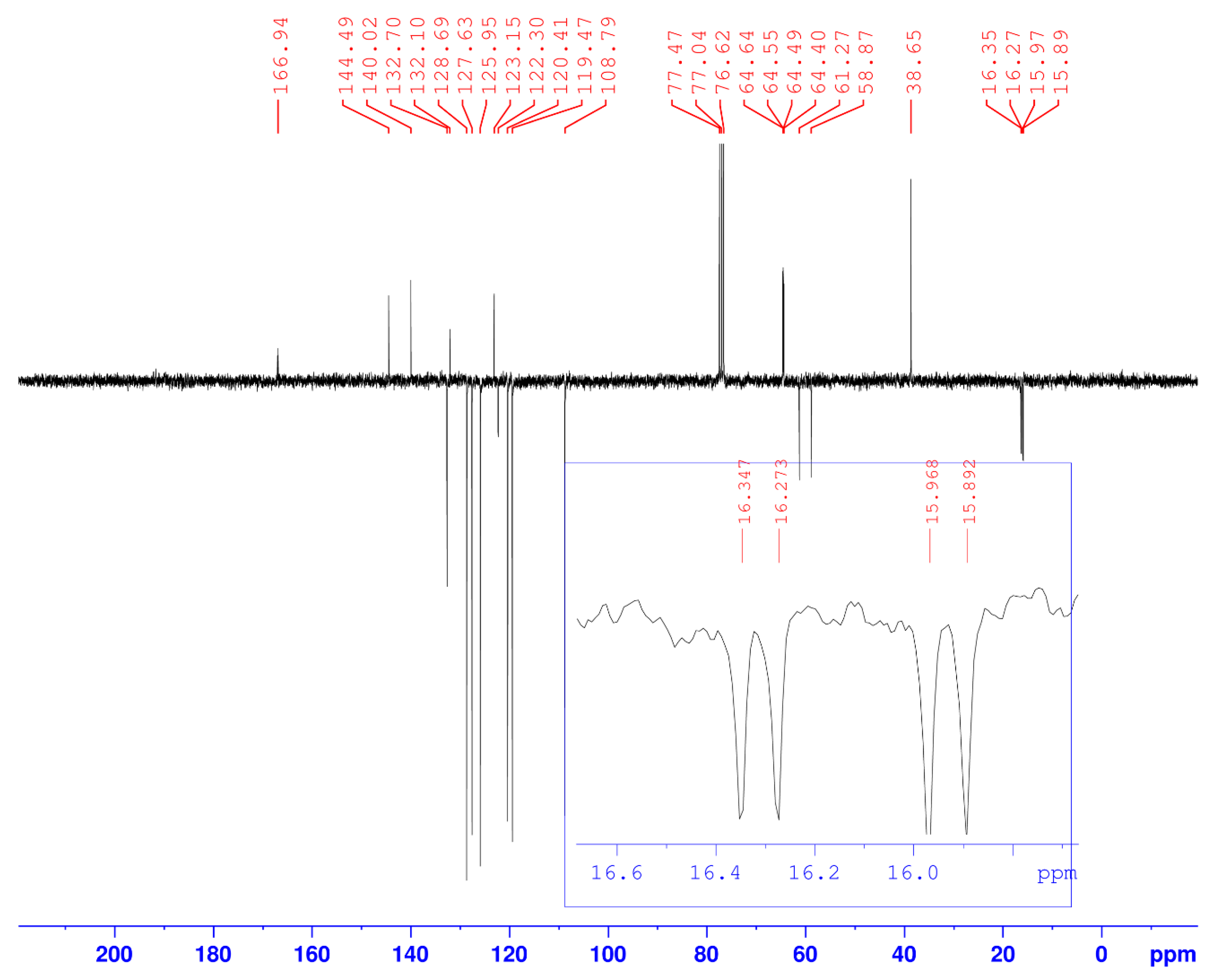
| 4 | - | 144.49 | - | - |
| 5 | 7.61 (s) | 122.30 | 1H5-1H5 | C5-1H5 |
| 6 | 5.65 (s) | 38.65 | 2H6-2H6 | C6-2H6 |
| 7 and 7′ | - | 140.42 | - | - |
| 8 and 8′ | 8.08–8.11 (d, J = 8) | 120.41 | 1H8-1H8 1H8′-1H8′ | C8-1H8 C8′-1H8′ |
| 9 and 9′ 13–18 | 7.40–7.55 (m) | 108.79 120.41–132.70 | 1H9-1H9; 1H9′-1H9′ 5H arom(Ph)-5Harom(Ph) | C9-1H9; C9′-1H9′ 5C arom(Ph)-5Harom(Ph) |
| 10 and 10′ | 7.22–7.28 (m) | 119.47 | 1H10-1H10 1H10′-1H10′ | C10-1H10 C10′-1H10′ |
| 11 and 11′ | 7.79–7.82 (d, J = 8) | 127.63 | 1H11-1H11 1H11′-1H11′ | C11-1H11 C11′-1H11′ |
| -NH- | 7.90–7.95 (dd, 3JH-H = 9.66; 3JH-P = 5.01) | - | 1Hamide-1Hamide 1Hamide-1H3 | - |
| 19 | - | 166.94 | - | - |
| Liaison | Vibration | Wavenumber (cm−1) |
|---|---|---|
| C–H | ν(Csp3-H) | 2850–3000 |
| δ(Csp3-H) | ~1400 | |
| C=C | ν(Csp2-Csp2) | 1500–1600 |
| C–Haromatic | ν(Csp2-H) | 3000–3100 |
| C–H | δ(Csp2-H) | ~985 and ~910 |
| N–Hamide | ν(N-H) | 3050–3500 |
| C=Oamide | ν(C=O) | 1650 |
| C–N | ν(C-N) | 1020–1220 |
| P=O | ν(P=O) | 1250 |
| P–O–C | ν(P-O-C) | 1050 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khadir Fall, S.A.; Achamlale, S.; Aouine, Y.; Nakkabi, A.; Faraj, H.; Alami, A. Diethyl [(4-{(9H-carbazol-9-yl)methyl}-1H-1,2,3-triazol-1-yl)(benzamido)methyl]phosphonate. Molbank 2020, 2020, M1167. https://doi.org/10.3390/M1167
Khadir Fall SA, Achamlale S, Aouine Y, Nakkabi A, Faraj H, Alami A. Diethyl [(4-{(9H-carbazol-9-yl)methyl}-1H-1,2,3-triazol-1-yl)(benzamido)methyl]phosphonate. Molbank. 2020; 2020(4):M1167. https://doi.org/10.3390/M1167
Chicago/Turabian StyleKhadir Fall, Serigne Abdou, Saïd Achamlale, Younas Aouine, Asmae Nakkabi, Hassane Faraj, and Anouar Alami. 2020. "Diethyl [(4-{(9H-carbazol-9-yl)methyl}-1H-1,2,3-triazol-1-yl)(benzamido)methyl]phosphonate" Molbank 2020, no. 4: M1167. https://doi.org/10.3390/M1167
APA StyleKhadir Fall, S. A., Achamlale, S., Aouine, Y., Nakkabi, A., Faraj, H., & Alami, A. (2020). Diethyl [(4-{(9H-carbazol-9-yl)methyl}-1H-1,2,3-triazol-1-yl)(benzamido)methyl]phosphonate. Molbank, 2020(4), M1167. https://doi.org/10.3390/M1167