3-Hydroxy-2-iodophenyl-(4-methylbenzenesulfonate)

 ,
, {kind=link}
Abstract
1. Introduction
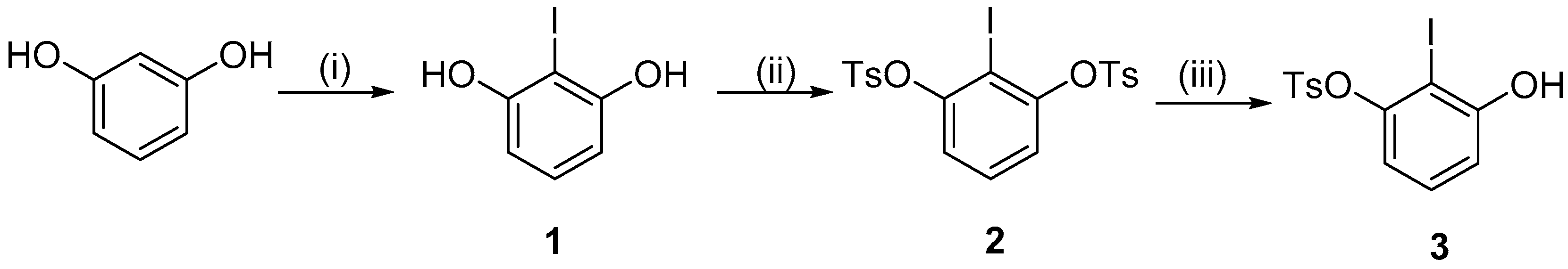
2. Results and Discussion
3. Materials and Methods
3.1. 2-Iodoresorcinol (1)
3.2. 2-Iodo-1,3-Phenylene Bis(4-Methylbenzenesulfonate) (2)
3.3. 3-Hydroxy-2-iodophenyl-(4-methylbenzenesulfonate) (3)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Allen, P.; Bragg, R.A.; Caffrey, M.; Ericsson, C.; Hickey, M.J.; Kingston, L.P.; Elmore, C.S. The synthesis of a tritium, carbon-14, and stable isotope-labeled cathepsin C inhibitors. J. Label Compd. Radiopharm. 2017, 60, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Steinhardt, R.C.; O’Neill, J.M.; Rathbun, C.M.; McCutcheon, D.C.; Paley, M.A.; Prescher, J.A. Design and synthesis of an alkynyl luciferin analogue for bioluminescence imaging. Chem. Eur. J. 2016, 22, 3671–3675. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Debnath, S.; Das, B. Synthesis of seven-membered fused sulfones by reductive Heck cyclization: An investigation for stereochemistry through DFT study. Tetrahedron 2015, 71, 476–486. [Google Scholar] [CrossRef]
- Garcia-Lopez, J.A.; Cetin, M.; Greaney, M.F. Synthesis of Hindered Biaryls via Aryne Addition and in Situ Dimerization. Org. Lett. 2015, 17, 2649–2651. [Google Scholar] [CrossRef] [PubMed]
- Mamiko, N.; Yoshio, A.; Fumitaka, K.; Ohmori, K.; Suzuki, K. Total synthesis of Actinorhodin. Angew. Chem. Int. Ed. 2019, 58, 4264–4270. [Google Scholar]
- Marsh, G.; Athanasiadou, M.; Athanassiadis, I.; Bergman, A.; Endo, T.; Haraguchi, K. Identification, quantification, and synthesis of a novel dimethoxylated polybrominated biphenyl in marine mammals caught off the coast of Japan. Environ. Sci. Technol. 2005, 39, 8684–8690. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Takiguchi, H.; Ohmori, K.; Suzuki, K. Total syntheses of pusilatins A–C, liverwort-derived macrocyclic bisbibenzyl dimers. Org. Lett. 2018, 20, 3579–3582. [Google Scholar] [CrossRef] [PubMed]
- Moreno, D.R.R.; Giorgi, G.; Salas, C.O.; Tapia, R.A. New short strategy for the synthesis of the dibenz [b,f] oxepin scaffold. Molecules 2013, 18, 14797–14806. [Google Scholar] [CrossRef] [PubMed]
- Tsujiyama, S.-I.; Suzuki, K. Preparation of benzocyclobutenone derivatives based on an efficient generation of benzynes. Org. Synth. 2007, 84, 272–284. [Google Scholar]
- Hamura, T.; Hosoya, T.; Yamaguchi, H.; Kuriyama, Y.; Tanabe, M.; Miyamoto, M.; Yasui, Y.; Matsumoto, T.; Suzuki, K. Facile Access to versatile polyaromatic building blocks: Selectively protected benzocyclobutenedione derivatives via regioselective [2 + 2] cycloaddition of α-alkoxybenzyne and ketene silyl acetal. Helv. Chim. Acta 2002, 85, 3589–3603. [Google Scholar] [CrossRef]
- Yoshida, S.; Morita, T.; Hosoya, T. Synthesis of diverse benzotriazoles from aryne precursors bearing an azido group via inter-and intramolecular cycloadditions. Chem. Lett. 2016, 45, 726–728. [Google Scholar] [CrossRef]
- Clark, C.G., Jr.; Floudas, G.A.; Lee, Y.J.; Graf, R.; Spiess, H.W.; Mullen, K. Molecularly tethered amphiphiles as 3-d supramolecular assembly platforms: Unlocking a trapped conformation. J. Am. Chem. Soc. 2009, 131, 8537–8547. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, F.; Guo, Y.; Shen, J.; Pan, X.; Hu, B.; Zheng, W.; Maddaluno, J.; Durandetti, M. 3-Hydroxy-2-iodophenyl-(4-methylbenzenesulfonate). Molbank 2020, 2020, M1158. https://doi.org/10.3390/M1158
Pan F, Guo Y, Shen J, Pan X, Hu B, Zheng W, Maddaluno J, Durandetti M. 3-Hydroxy-2-iodophenyl-(4-methylbenzenesulfonate). Molbank. 2020; 2020(4):M1158. https://doi.org/10.3390/M1158
Chicago/Turabian StylePan, Feng, Yi Guo, Jinying Shen, Xiaofeng Pan, Binbin Hu, Weixin Zheng, Jacques Maddaluno, and Muriel Durandetti. 2020. "3-Hydroxy-2-iodophenyl-(4-methylbenzenesulfonate)" Molbank 2020, no. 4: M1158. https://doi.org/10.3390/M1158
APA StylePan, F., Guo, Y., Shen, J., Pan, X., Hu, B., Zheng, W., Maddaluno, J., & Durandetti, M. (2020). 3-Hydroxy-2-iodophenyl-(4-methylbenzenesulfonate). Molbank, 2020(4), M1158. https://doi.org/10.3390/M1158