2-(Fluoromethyl)-4,7-dimethoxy-1-methyl-1H-benzimidazole




Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
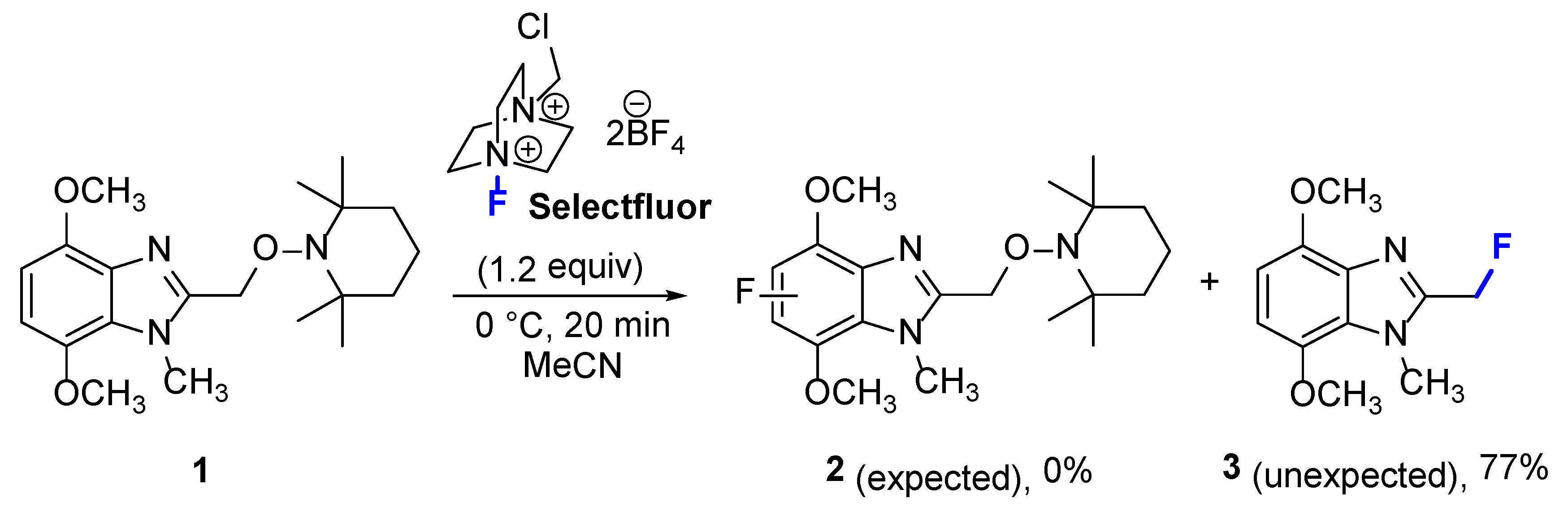
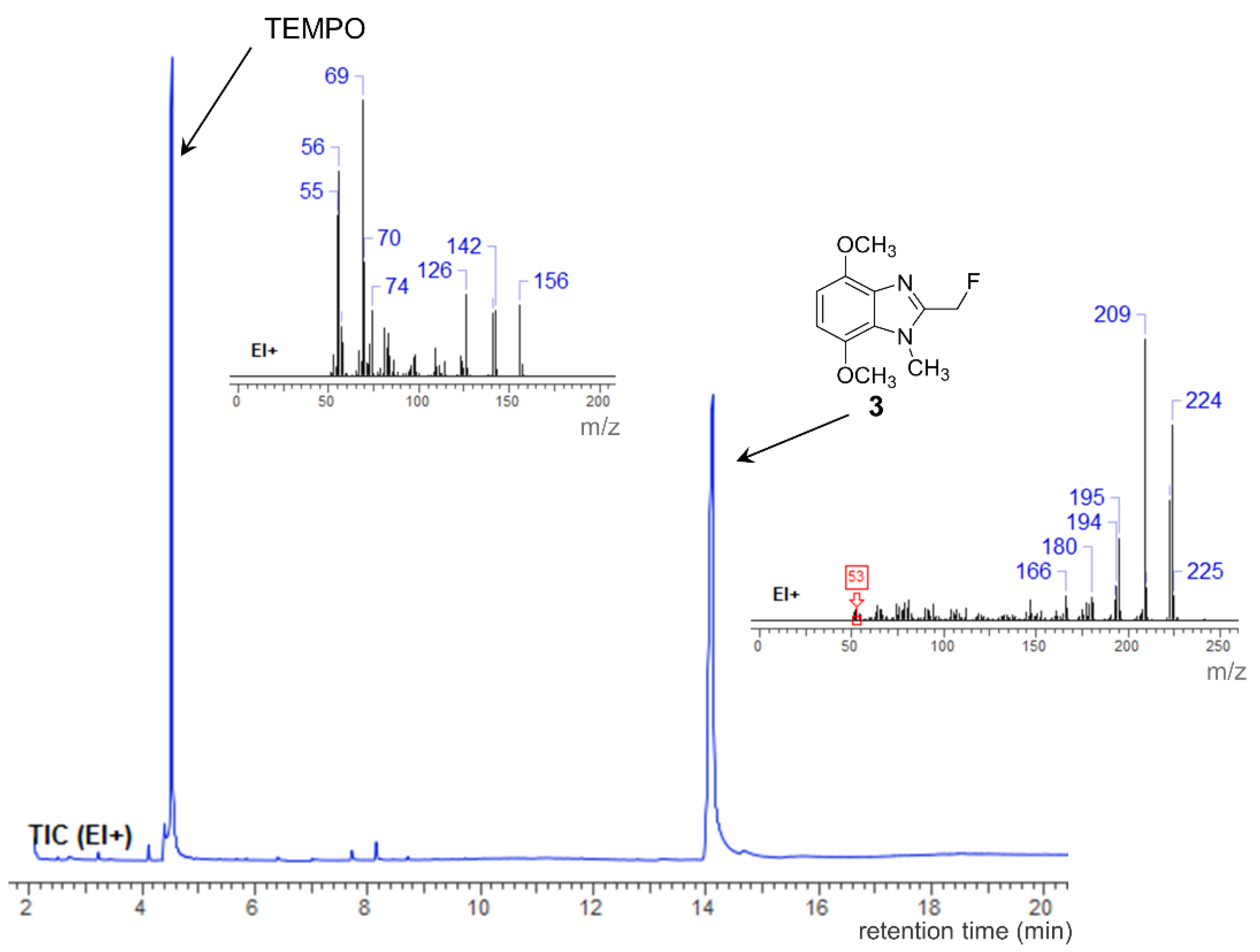
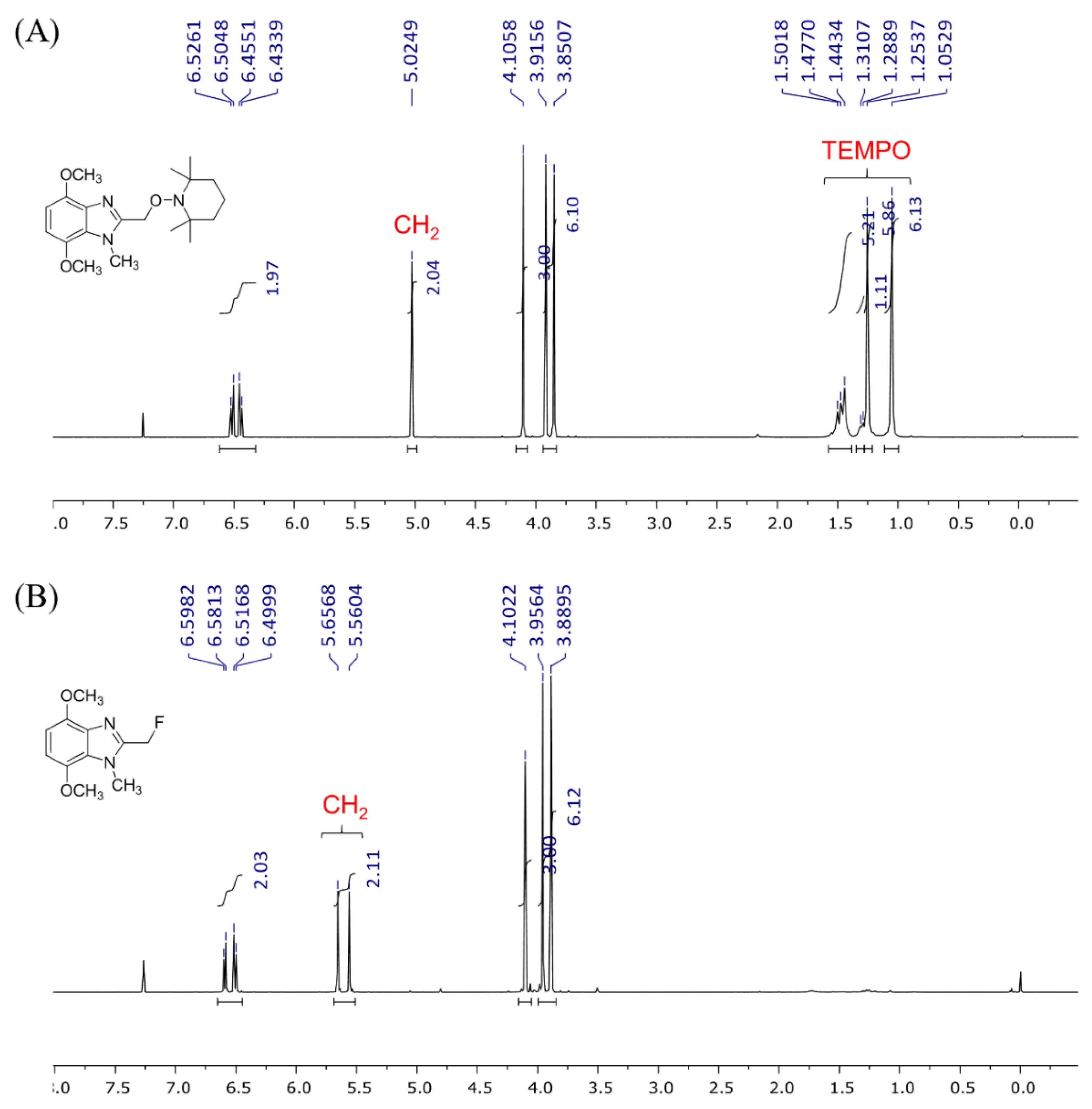
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Measurements
3.2. Synthesis of 2-(Fluoromethyl)-4,7-dimethoxy-1-methyl-1H-benzimidazole (3)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nicolas, J.; Guillaneuf, Y.; Lefay, C.; Bertin, D.; Gigmes, D.; Charleux, B. Nitroxide-mediated polymerization. Prog. Polym. Sci. 2013, 38, 63–235. [Google Scholar] [CrossRef]
- Kielty, P.; Farràs, P.; McArdle, P.; Smith, D.A.; Aldabbagh, F. Visible-light unmasking of heterocyclic quinone methide radicals from alkoxyamines. Chem. Commun. 2019, 55, 14665–14668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyffeler, P.T.; Durόn, S.G.; Burkart, M.D.; Vincent, S.P.; Wong, C.-H. Selectfluor: Mechanistic Insight and applications. Angew. Chem. Int. Ed. 2005, 44, 192–212. [Google Scholar] [CrossRef] [PubMed]
- Banks, R.E.; Besheesh, M.K.; Mohialdin-Khaffaf, S.N.; Sharif, I. N-Halogeno compounds. Part 18. 1-Alkyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane salts: User-friendly site-selective electrophilic fluorinating agents of the N-fluoroammonium class. J. Chem. Soc. Perkin Trans. 1 1996, 16, 2069–2076. [Google Scholar] [CrossRef]
- Heravi, M.R.P. Fluorination of activated aromatic systems with Selectfluor F-TEDA-BF4 in ionic liquids. J. Fluorine Chem. 2008, 129, 217–221. [Google Scholar] [CrossRef]
- Liang, D.; Li, Y.; Gao, S.; Li, R.; Li, X.; Wang, B.; Yang, H. Amide-assisted radical strategy: Metal-free direct fluorination of arenes in aqueous media. Green Chem. 2017, 19, 3344–3349. [Google Scholar] [CrossRef]
- Mirallai, S.I.; Koutentis, P.A.; Aldabbagh, F. Regioselective fluorination of 7-oxo-1,2,4-benzotriazines using Selectfluor. Molecules 2019, 24, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- René, O.; Souverneva, A.; Magnuson, S.R.; Fauber, B.P. Efficient syntheses of 2-fluoroalkylbenzimidazoles and –benzothiazoles. Tetrahedron Lett. 2013, 54, 201–204. [Google Scholar] [CrossRef]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of fluorine and fluorine-containing functional groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammill, C.L.; Noble, B.B.; Norcott, P.L.; Ciampi, S.; Coote, M.L. Effect of Chemical Structure on the Electrochemical Cleavage of Alkoxyamines. J. Phys. Chem. C 2019, 123, 5273–5281. [Google Scholar] [CrossRef] [Green Version]
- Aldabbagh, F.; Busfield, W.K.; Jenkins, I.D.; Thang, S.H. The reactivity of nitroxides towards alkenes. Tetrahedron Lett. 2000, 41, 3673–3676. [Google Scholar] [CrossRef]
- Kielty, P. Heterocyclic Chemistry: Controlled Unmasking of Nitric Oxide and Nitroxides. Ph.D. Thesis, National University of Ireland Galway, Galway City, Ireland, 2019. [Google Scholar]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kielty, P.; Farràs, P.; Smith, D.A.; Aldabbagh, F. 2-(Fluoromethyl)-4,7-dimethoxy-1-methyl-1H-benzimidazole. Molbank 2020, 2020, M1129. https://doi.org/10.3390/M1129
Kielty P, Farràs P, Smith DA, Aldabbagh F. 2-(Fluoromethyl)-4,7-dimethoxy-1-methyl-1H-benzimidazole. Molbank. 2020; 2020(2):M1129. https://doi.org/10.3390/M1129
Chicago/Turabian StyleKielty, Patrick, Pau Farràs, Dennis A. Smith, and Fawaz Aldabbagh. 2020. "2-(Fluoromethyl)-4,7-dimethoxy-1-methyl-1H-benzimidazole" Molbank 2020, no. 2: M1129. https://doi.org/10.3390/M1129
APA StyleKielty, P., Farràs, P., Smith, D. A., & Aldabbagh, F. (2020). 2-(Fluoromethyl)-4,7-dimethoxy-1-methyl-1H-benzimidazole. Molbank, 2020(2), M1129. https://doi.org/10.3390/M1129