2. Results and Discussion
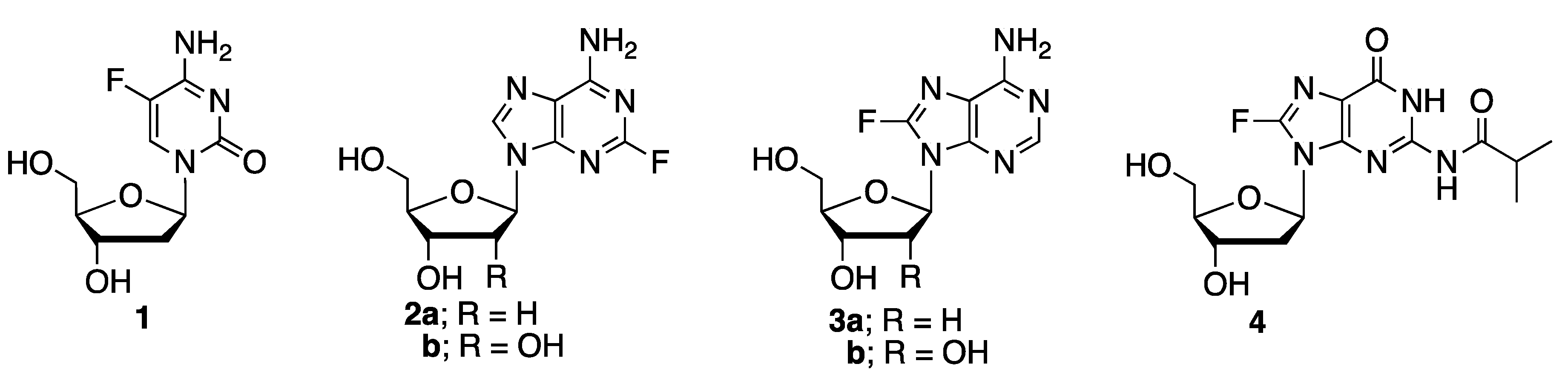
Syntheses of adenine nucleosides, such as 2-fluoro-2′-deoxyadenosine
2a [
4,
5,
6,
7] and 2-fluoroadenosine
2b [
6,
8] have been demonstrated through different approaches. Introduction of fluorine to the C-8 position of purine nucleosides is more challenging, likely due to the instability of the resulting nucleosides. The F-C bond in 8-F-rA
3b is labile under basic conditions. In this respect, basic conditions that are required for the removal of
N-acyl protecting groups can lead to defluorination. Thus, an early attempt to synthesize 8-fluoroadenosine
3b by the treatment of 2′,3′,5′-
O-triacetyl-8-fluoroadenosine with methanolic ammonia [
9] was later found not to give the desired 8-F-rA [
10]. Enzymatic [
11] or deprotection conditions under mild acidic conditions [
12] allowed for the access of 8-F-rA. Fluorination of guanosine at the C-8 position was demonstrated by treatment with elemental fluorine [
13]. This method, however, is inconvenient due to the difficulty in handling elemental fluorine. More recently, syntheses of protected 8-fluoro deoxyinosine, deoxyadenosine (which led to the formation of 8-fluoro-2′-deoxyadenosine
3a after deprotection), and deoxyguanosine derivatives were demonstrated through metalation-electrophilic fluorination by the treatment of suitably protected purine nucleosides with lithium diisopropylamide (LDA) followed by
N-fluorobenzenesulfonimide (NFSI) [
14], all in low or moderate yields. In this chemistry, guanosine derivatives had to be protected at
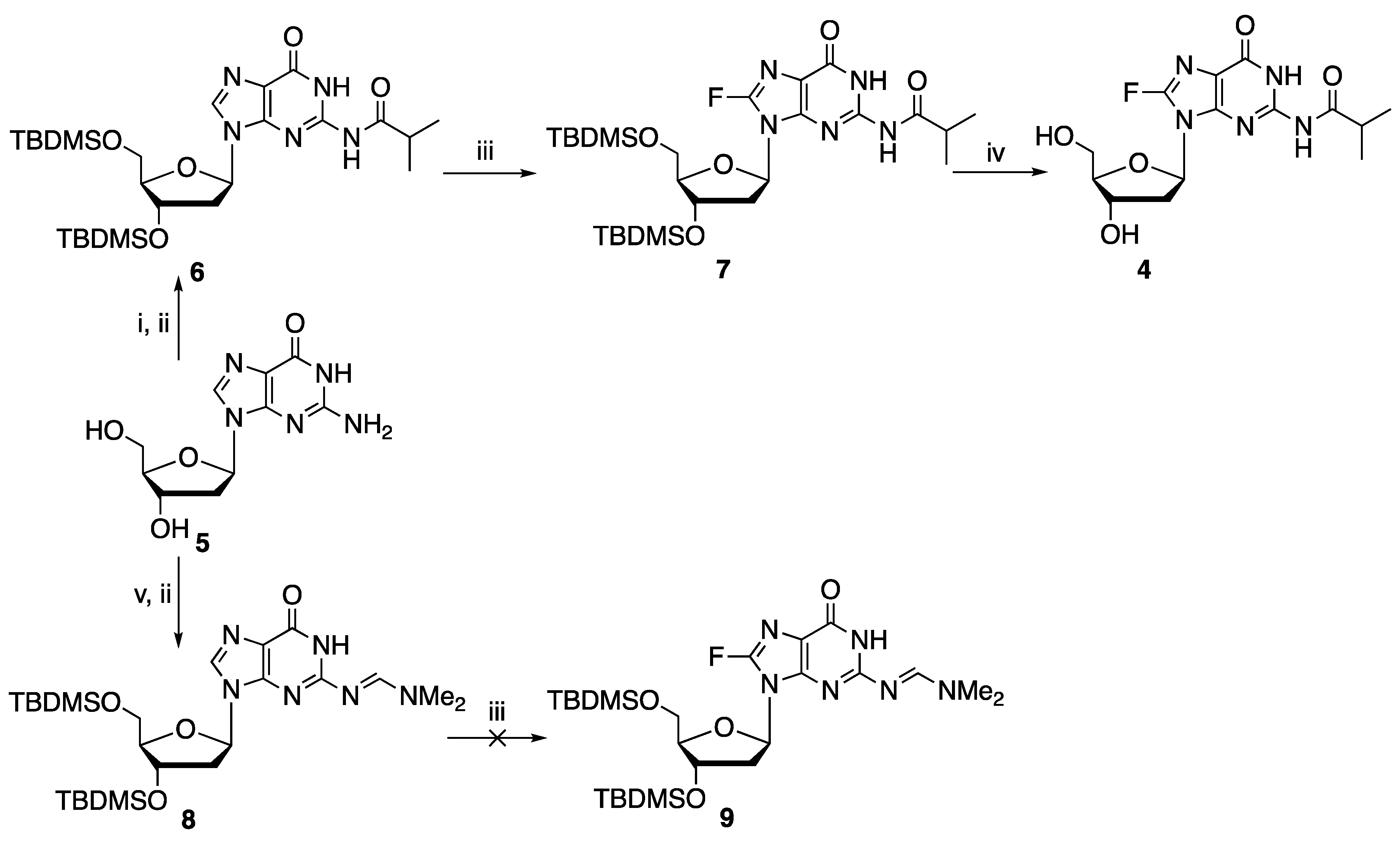
O-6 position. We found that
N-2-isobutyryl protected, but not
N-2-formamidine protected, 2′-deoxyguanosine, compounds
6 and
8, respectively, can be fluorinated using this chemistry. Thus, 3′,5′-
O-bis(
tert-butyldimethylsilyl)-
N-2-isobutyryl-2′-deoxyguanosine
6 was subjected to metalation−electrophilic fluorination with LDA and NSFI to generate the corresponding 8-fluoro analog
7 in 30% yield (
Scheme 1), with recovery of 50% the starting material
6. A similar reaction with 3′,5′-
O-bis(
tert-butyldimethylsilyl)-
N-2- dimethylformamidine-2′-deoxyguanosine
8 failed to give the corresponding fluorinated product
9.
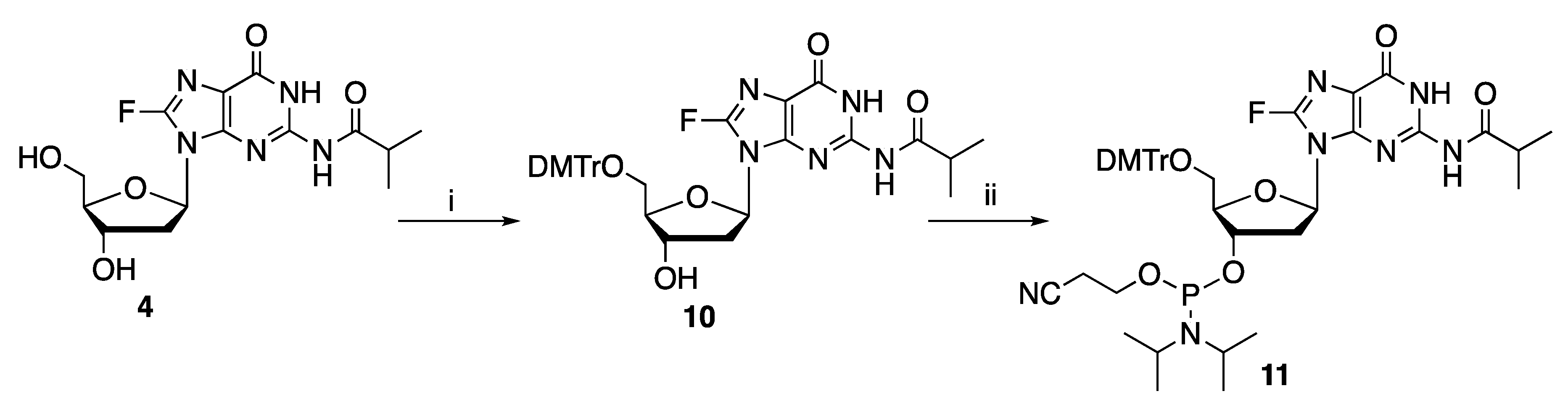
In order to introduce 8-fluoro-2′-deoxyguanosine into oligonucleotides through the phosphoramidite chemistry-based solid phase synthesis, the stability of 8-fluoro-N-2-isobutyryl-2′-deoxyguanosine 12 in acidic conditions was investigated. This property is of importance as di- or trichloroacetic acid (DCA and TCA, respectively) is typically used for the removal of dimethoxytrityl (DMTr) group during the detritylation reaction. As such, 8-fluoro-2′-deoxyguanosine derivatives will need to be sufficiently stable under the acidic detritylation condition. Thus, experiments were conducted by 19F NMR spectroscopy to determine the stability of 8-fluoro-N-isobutyryl-2′-deoxyguanosine 4 in acids of different pKa.
As seen in
Table 1, 8-fluoro-
N-isobutyryl-2′-deoxyguanosine
4 is stable in 3% benzoic acid and 80% acetic acid in methanol, and relatively stable in monochloroacetic acid (MCA), however, removal of the DMTr group with these acids is reversible. While 8-fluoro-
N-isobutyryl-2′-deoxyguanosine
4 is relatively unstable in DCA and TCA, It was previously shown that DMTr removal can be effected by DCA and TCA in as little as 20 s [
15], thus, DCA appears to be the most suited for the removal of DMTr groups in solid phase synthesis involving 8-fluoro-
N-isobutyryl-2′-deoxyguanosine
4. This nucleoside was subsequently protected with DMTr at 5′-OH (as in
10) and then transformed into the corresponding 3′-phosphoramidite
11 (
Scheme 2).
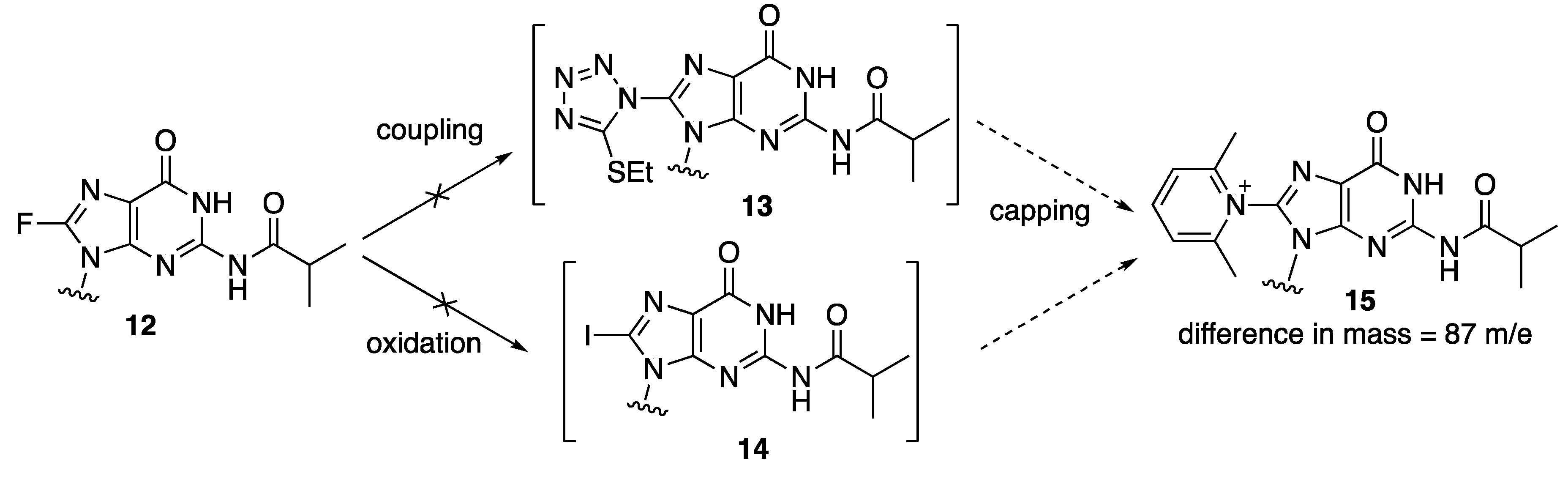
Solid phase syntheses were carried out using the ABI standard 1 μmol cycle conditions for the assembly of d(CG)
6 sequences where a single dG was replaced with 8-fluoro-dG at each of the dG positions. The products were cleaved from the solid support and deprotected by incubation with concentrated aqueous ammonium hydroxide at 55 °C overnight. All modified sequences were found by mass spectrometry to contain a major species with a mass of 3753 Da, which is 87 mass unit higher than the expected sequences (3665 Da). The exact nature of the modification during solid phase synthesis that led to the formation of this higher mass species remains to be identified. It was found that the treatment of 8-fluoro-
N-2-isobutyryl-2′-deoxyguanosine
12 with the activator (5-ethylthio-1
H-tetrazole) for coupling reactions or the oxidation solution containing aqueous iodine in pyridine, did not lead to the formation of displacement products
13 and
14, respectively, which, if reacted with 2,6-lutidine during the capping step, could generate the corresponding modified dG species
15 with the 87 extra mass units found in the oligonucleotides (
Figure 2).
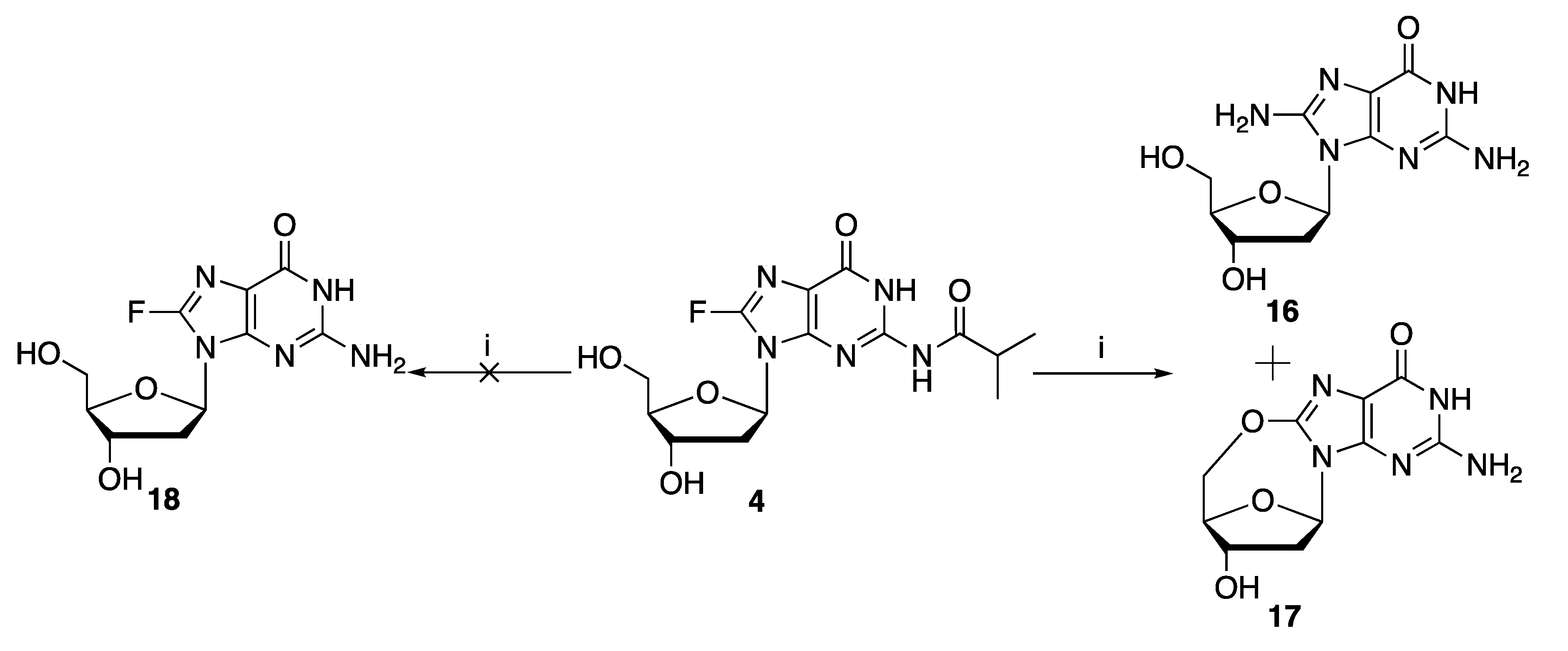
Finally, it was found that treatment of 8-fluoro-
N-isobutyryl-2′-deoxyguanosine
4 (
Scheme 3) with concentrated aqueous ammonium hydroxide at 55 °C overnight, the typical conditions for the deprotection of
N-acyl groups, did not give the desired 8-fluoro-2′-deoxyguanosine
18; instead, a mixture of 8-amino-2′-deoxyguanosine
16 and C8:5′-
O-cyclo-2′-deoxyguanosine
17 was obtained in approximately a 2:1 ratio, as indicated by the reverse phase HPLC profile in
Figure S28.
Taken together, these results suggest that 8-fluoro-N-isobutyryl-2′-deoxyguanosine 4 is unsuitable for the introduction of 8-fluoro-2′-deoxyguanosine into oligonucleotides via the phosphoramidite chemistry-based solid phase synthesis. Alternative N2-protecting groups, as well as modified solid phase synthesis conditions, will be required for successful incorporation of 8-fluoro-2′-deoxyguanosine into oligonucleotides.
3. Materials and Methods
1H NMR spectra were measured at 400 MHz with a Bruker Avance 400 Digital NMR spectrometer (Billerica, MA, USA). 13C, 31P and 19F NMR spectra were recorded at 100.6, 162.0 and 376.6 MHz respectively, with the same spectrometer. Chemical shifts and coupling constants (J values) are given in ppm and Hz, respectively. Deuterated solvents were purchased from C/D/N Isotopes (Montreal, QC, Canada). EI (electron impact) and FAB (fast atom bombardment) mass spectra were obtained with a Thermo Scientific DFS mass spectrometer (Waltham, MA, USA); ESI (electrospray) spectra were measured with a Bruker HCT Plus ion-trap mass spectrometer. Desican silica gel (230–400 mesh) was used for column chromatography. Thin layer chromatography was performed on Silicycle F-254 silica TLC plates using the following solvent mixtures:
Solvent A: methanol-dichloromethane (5:95, v/v)
Solvent B: methanol-dichloromethane (20:80, v/v)
3.1.Solvents and Chemicals
Toluene was dried by heating under reflux over sodium in the presence of benzophenone for 4 h and then distilled under nitrogen. N,N-Diisopropylethylamine and pyridine were dried by heating under reflux over calcium hydride for 4 h and then distilled under nitrogen. N,N-Dimethylformamide was dried by heating at 60 °C over calcium hydride for 4 h and then distilled under vacuum. Dichloromethane was dried by heating under reflux over phosphorus pentoxide for 4 h and then distilled under nitrogen. All other reagents were purchased from Sigma-Aldrich or TCI America without further purification prior to use unless stated otherwise.
3.2. 3′,5′-Bis(tert-butyldimethylsilyl)-N-isobutyryl-2′-deoxyguanosine (6)
N-Isobutyryl-2′-deoxyguanosine 5 (3.50 g, 10.4 mmol), imidazole (4.66 g, 68.6 mmol) and tert-butyldimethylsilyl chloride (6.90 g, 45.7 mmol) were stirred in dry dimethylformamide (30 mL) for 5 h. The resulting slurry was diluted with ethanol (25 mL) and stored at −20 °C for 24 h. White crystals were collected by vacuum filtration to give the title compound (4.93 g, 84%). M.p. 102–104 °C (ethanol). Rf (system A): 0.38. HR-MS (EI) found 565.3099, C26H47N5O5Si2 required 565.3116. δH (CDCl3): 0.09, 0.10, 0.117 and 0.119 (total 12 H, Si(CH3)2), 0.920 and 0.925 (total 18 H, Si(C(CH3)3), 1.27 (3 H, d, J = 6.5, iBu), 1.29 (3 H, d, J = 6.5), 2.37 (1 H, ddd, J = 3.8, 6.0 and 10.0, H-2′), 2.47 (1 H, m, H-2′’), 2.65 (1 H, CH(CH3)2, septet, J = 6.9), 3.78 (2 H, m, H-5′/5′’), 3.99 (1 H, q, J = 3.4, H-4′), 4.58 (1 H, m, H-3′), 6.23 (1 H, dd, J = 6.5 and 6.5, H-1′), 7.98 (1 H, s, H-8), 8.40 (1 H, s, NH), 12.00 (1 H, s, NH). δC (CDCl3): −5.51, −5.38, −4.77, −4.68, 18.0, 18.4, 18.95, 19.01, 25.7, 26.0, 36.6, 41.4, 62.8, 71.8, 83.5, 88.0, 121.5, 136.8, 147.3, 147.8, 155.5, 178.1.
3.3. 3′,5′-Bis(tert-butyldimethylsilyl)-8-fluoro-N-isobutyryl-2′-deoxyguanosine (7)
3′,5′-Bis(tert-butyldimethylsilyl)-N-isobutyryl-2′-deoxyguanosine 6 (4.00 g, 7.08 mmol) was co-evaporated with dry toluene (3 × 15 mL) and dissolved in dry toluene (80 mL). After the mixture was cooled to −78 °C (acetone–dry ice bath), 2.0 M lithium diisopropylamide solution in THF/heptane/ethylbenzene (17.7 mL, 35.4 mmol) was added slowly. After 2 h, N-fluorobenzenesulphonamide (6.68 g, 21.2 mmol) was added and the mixture was stirred for 1.5 h at −78 °C and then allowed to warm up to room temperature over 30 min. Saturated aqueous ammonium chloride solution (80 mL) was added and the layers were separated. The aqueous layer was extracted with dichloromethane (2 × 40 mL), and the combined organic layers were washed with saturated sodium bicarbonate solution (100 mL), dried over magnesium sulfate and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel. The appropriate fractions, which were eluted with dichloromethane–methanol (99.3:0.7 v/v), were pooled and concentrated under reduced pressure to give the title compound as a pale yellow solid (825 mg, 20%). Rf (system A): 0.41. HR-MS (EI) found 583.3015, C26H46N5O5Si2F required 583.3022. δH (CDCl3): 0.04 (3 H, s, Si(CH3)2), 0.06 (3 H, s, Si(CH3)2), 0.13 (6 H, s, Si(CH3)2), 0.89 (9 H, s, Si(C(CH3)3)2), 0.93 (9 H, s, Si(C(CH3)3)2), 1.24 (3 H, d, J = 7.0, CH(CH3)2), 1.27 (3 H, d, J = 7.0, CH(CH3)2), 2.26 (1 H, ddd, J = 3.9, 6.6 and 10.7, H-2′), 2.66 (1 H, CH(CH3)2, septet, J = 6.9), 2.78 (1 H, m, H-2′’), 3.71 (2 H, m, H-5′/5′’), 3.89 (1 H, m, H-4′), 4.59 (1 H, m, H-3′), 6.16 (1 H, dd, J = 6.8 and 6.8, H-1′), 8.69 (1 H, s, NH), 12.12 (1 H, s, NH). δF (CDCl3): −101.3. δC (CDCl3): −5.5, −5.4, −4.8, −4.7, 18.0, 18.4, 18.8, 19.1, 25.8, 25.9, 36.5, 37.9, 62.7, 71.7, 82.3, 87.7, 114.8 (d, JC,F = 13.9), 146.7 (d, JC,F = 3.3), 147.7 (d, JC,F = 2.7), 149.6 (d, JC,F = 251.1), 154.8, 178.5.
3.4. 8-Fluoro-N-isobutyryl-2′-deoxyguanosine (4)
3′,5′-Bis(tert-butyldimethylsilyl)-8-fluoro-N-isobutyryl-2′-deoxyguanosine 7 (350 mg, 0.60 mmol) and tetra-n-butylammonium fluoride (391 mg, 1.5 mmol) were stirred in tetrahydrofuran (12 mL) for 1 h at r.t., followed by addition of warm water (50 °C) until all salts dissolved. The product crystalized from water to give 127 mg (60%) of the title compound as white crystals. M.p.: decomposed at 183–185 °C (water). Rf (system B): 0.72. HR-MS (EI) found 335.1220, C14H18N5O5F requires 355.1292. δH (DMSO-d6): 1.13 (6 H, d, J = 6.6, CH(CH3)2), 2.24 (1 H, ddd, J = 3.8, 6.5 and 10.2, H-2′), 2.76 (2 H, m, H-2′’ and CH(CH3)2), 3.79 (1 H, m, H-4′), 4.37 (1 H, m, H-3′), 4.84 (1 H, t, J = 5.6, 5′-OH, ex), 5.35 (1 H, d, J = 4.0, 3′-OH, ex), 6.17 (1 H, t, J = 6.9, H-1′), 11.68 (1 H, s, NH, ex), 12.17 (1 H, s, NH, ex). δC (DMSO-d6): 14.0, 19.3, 35.3, 37.5, 62.0, 70.7, 82.8, 88.2, 113.9 (d, JC,F = 13.1), 147.5 (d, JC,F = 3.2), 148.9 (d, JC,F = 2.6), 149.5 (d, JC,F = 247.6), 154.4 (d, JC,F = 2.1), 180.6. δF (DMSO-d6): −102.9.
3.5. 5′-(4,4′-Dimethoxytrityl)-8-fluoro-N-isobutyryl-2′-deoxyguanosine (10)
8-Fluoro-N-isobutyryl-2′-deoxyguanosine 4 (0.50 g, 1.4 mmol) was co-evaporated with dry pyridine (3 × 10 mL) and dissolved in dry pyridine (5 mL), followed by addition of 4,4′-dimethoxytrityl chloride (0.72 g, 2.1 mmol). After 30 min, the reaction was quenched with a triethylamine/ethanol (1:1 v/v) mixture (1 mL). The products were diluted with dichloromethane (50 mL) and extracted with saturated aqueous sodium hydrogen carbonate (50 mL). The aqueous layer was back-extracted with dichloromethane (30 mL) and the combined organic layers were dried (MgSO4) and evaporated under reduced pressure. The oily residue was purified by column chromatography on silica gel. The appropriate fractions, which were eluted with dichloromethane–ethanol–triethylamine (98.2:1.5:0.3 v/v/v), were pooled and concentrated under reduced pressure to give the title compound as a colorless glass (850 mg, 92%). Rf (system A): 0.27. MS (ESI) found 658.2 (M-H+), C35H37FN5O7+ required 658.2677. δH (CDCl3): 0.98 (3 H, d, J = 6.7), 1.07 (3 H, d, J = 7.0), 2.27 (1 H, CH(CH3)2, m), 2.34 (1 H, ddd, J = 4.2, 6.2 and 10.6, H-2′), 3.00 (1 H, m, H-2′’), 3.24 (1 H, dd, J = 5.2 and 10.3, H-5′), 3.36 (1 H, dd, J = 3.7 and 10.3, H-5′’), 3.74 and 3.75 (6 H, (OCH3)2), 4.09 (1 H, ddd, J = 4.1, 4.1 and 8.5, H-4′), 4.82 (1 H, m, H-3′), 6.14 (1 H, dd, J = 6.2 and 6.2, H-1′), 6.74 (3 H, d, J = 3.1), 6.77 (3 H, d, J = 3.1), 7.14–7.23 (3 H, m), 7.30 and 7.32 (4 H), 7.44 (2 H, dd, J = 1.5 and 8.3). δF(CDCl3): −103.7. δC (CDCl3): 18.75, 18.77, 36.0, 36.2, 55.2, 63.9, 71.4, 82.8, 86.1, 86.3, 113.1, 115.0, 126.9, 127.8, 128.1, 129.96, 130.0, 135.9, 136.1, 144.9, 146.9, 147.8, 148.6, 158.5, 179.4.
3.6. 5′-(4,4′-Dimethoxytrityl)-8-fluoro-N-isobutyryl-2′-deoxyguanosine-3′-(2-cyanoethyl)-N,N-diisopropylphosphoramidite (11)
5′-(4,4′-Dimethoxytrityl)-8-fluoro-N-isobutyryl-2′-deoxyguanosine 10 (1.00 g, 1.52 mmol) was co-evaporated with dry toluene (3 × 10 mL) and then dissolved in dry dichloromethane (20 mL), followed by addition of dry N,N-diisopropylethylamine (1.0 mL, 5.7 mmol) and (2-cyanoethyl)-N,N-diisopropyl phosphochloridite (0.72 g, 3.0 mmol). After the reaction mixture was stirred for 30 min, triethylamine (1 mL) was added and products were concentrated under reduced pressure. The residue was purified by column chromatography on silica gel. The appropriate fractions, which were eluted with hexane–acetone–triethylamine (70:28:2 v/v/v), were pooled and concentrated under reduced pressure. The resulting oil was dissolved in dichloromethane (2 mL) and added dropwise to pentane (20 mL) at −40 °C under stirring. The cloudy suspension was centrifuged for 20 min and the supernatant was decanted. The white solid was collected and dried to give the title phosphoramidite (0.57 g, 44%). HR-MS (FAB) found 858.3787 ([M + H]+), C44H54FN7O8P+ required 858.3755. δH(CDCl3) include the following signals: 2.45–2.56 (1 H, m, H-2′), 3.12–3.51 (3 H, m, H-2”, H-5′, and H-5”), 3.774, 3.777, 3.782 and 3.787 (6 H, -OMe), 4.21–4.29 (1 H, m, H-4′), 4.77–4.89 (1 H, m, H-3′), 6.19 (1 H, m, H-1′), 6.75–6.79 (4 H, m). δF (CDCl3): −106.27 and −104.28. δP(CDCl3): 147.78 and 147.96.
3.7. Treatment of 8-Fluoro-N-isobutyryl-2′-deoxyguanosine under Ammonolysis Conditions
8-Fluoro-N-isobutyryl-2′-deoxyguanosine (4 mg) was incubated with concentrated aqueous ammonium hydroxide (1.0 mL) at 55 °C for 16 h. Upon cooling the products were lyophilized. The residue was purified by column chromatography using C18-reverse phase matrix (1 × 15 cm) on a BioRad DuoFlow FPLC system. The column was eluted with acetonitrile–water (0:100 to 30:70 v/v) in a linear gradient over 60 min. Eluants were collected in 1.0 mL fractions and lyophilized.
3.8. Oligonucleotide Synthesis
Oligonucleotides were synthesized using an ABI 3400 DNA synthesizer (Waltham, MA, USA) under the standard 1 µmol cycle conditions. Phosphoramidites were prepared as 100 mM solutions in dry acetonitrile. A solution of 5-(ethylthio)-1H-tetrazole (250 mM in dry acetonitrile) was used as the activator, and coupling reaction time was set at 60 s. Detritylation was effected by delivering dichloroacetic acid (3% in dichloromethane) to reaction columns for 110 s. After solid phase synthesis was complete, the products were cleaved from solid support and deprotected by incubation with ammonium hydroxide at 55 °C for 24 h and then lyophilized.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}