6-Imino-1,2,3,4,8,9,10,11-octahydropyrido[1,2-a]pyrido[1′,2′:1,2]imidazo[4,5-f]benzimidazole-13-one: Synthesis and Cytotoxicity Evaluation



{kind=link}
{kind=link}
{kind=link}
Abstract
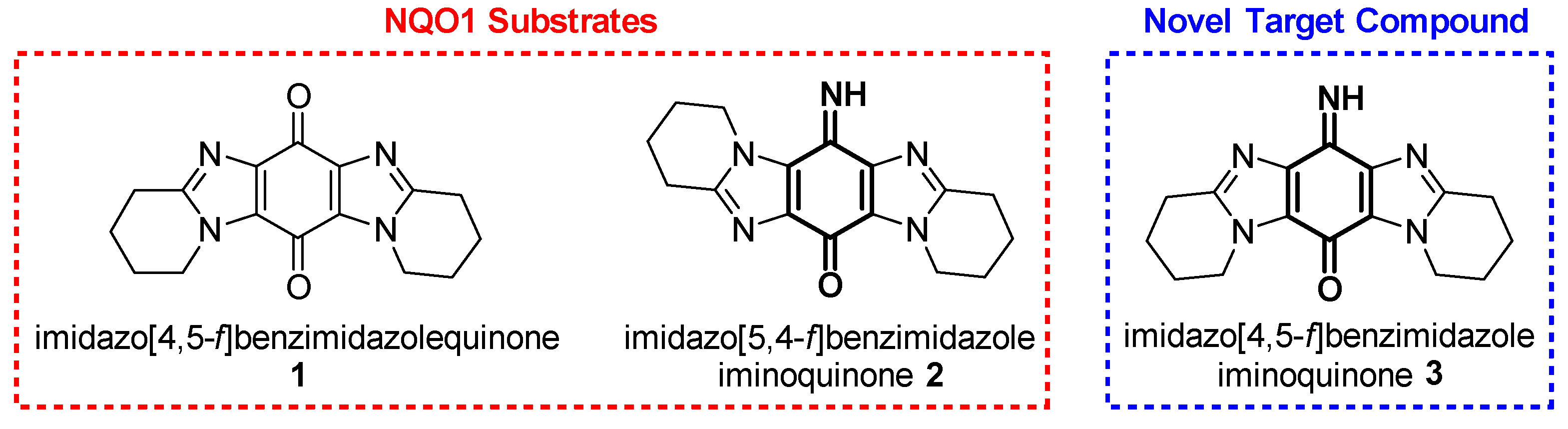
1. Introduction
2. Results and Discussion
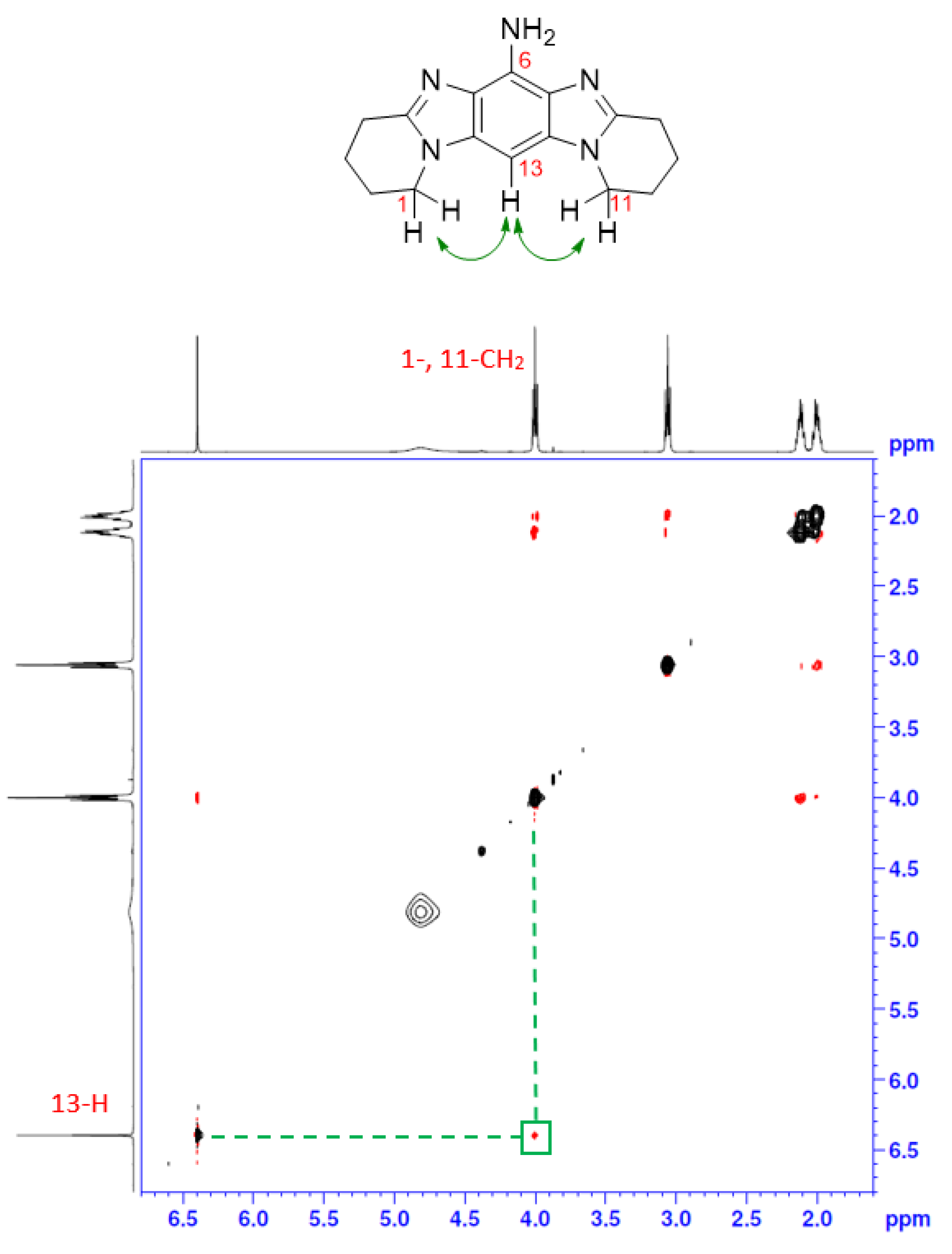
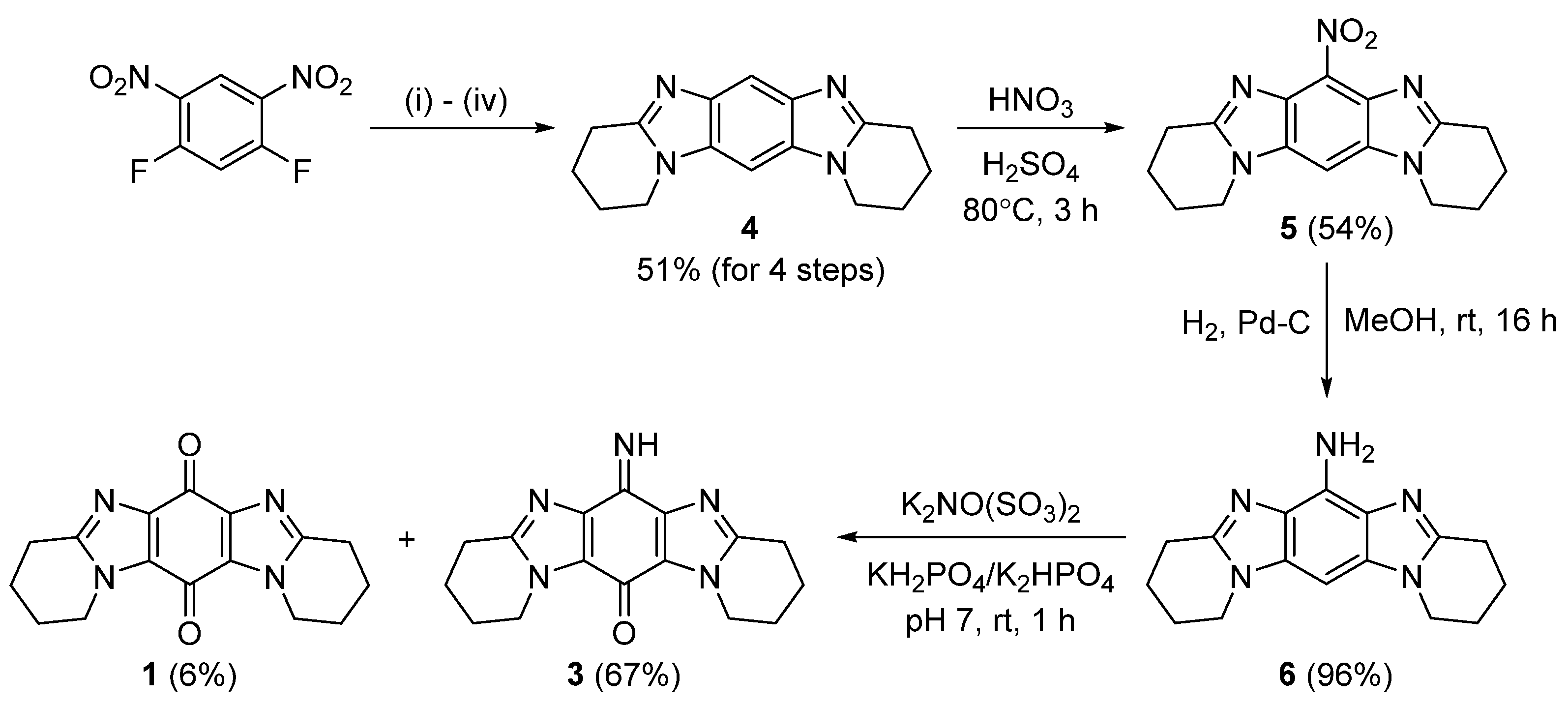
2.1. Synthesis
2.2. Cytotoxicity
3. Materials and Methods
3.1. Materials and Measurements
3.2. Synthetic Procedures
3.2.1. Synthesis of 1,2,3,4,8,9,10,11-Octahydropyrido[1,2-a]pyrido[1’,2’:1,2]imidazo[4,5-f]benzimidazole (4)
3.2.2. Synthesis of 6-Nitro-1,2,3,4,8,9,10,11-octahydropyrido[1,2-a]pyrido[1’,2’:1,2]imidazo[4,5-f]benzimidazole (5)
3.2.3. Syntheis of 1,2,3,4,8,9,10,11-Octahydropyrido[1,2-a]pyrido[1’,2’:1,2]imidazo[4,5-f]benzimidazol-6-amine (6)
3.2.4. Frémy Oxidation of 6 to Give Title Compound 3
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, K.; Chen, D.; Ma, K.; Wu, X.; Hao, H.; Jiang, S. NAD(P)H:quinone oxidoreductase 1 (NQO1) as a therapeutic and diagnostic target in cancer. J. Med. Chem. 2018, 61, 6983–7003. [Google Scholar] [CrossRef] [PubMed]
- Schulz, W.G.; Skibo, E.B. Inhibitors of topoisomerase II based on the benzodiimidazole and dipyrroloimidazobenzimidazole ring systems: Controlling DT-diaphorase reductive inactivation with steric bulk. J. Med. Chem. 2000, 43, 629–638. [Google Scholar] [CrossRef]
- Suleman, A.; Skibo, E.B. A comprehensive study of the active site residues of DT-diaphorase: Rational design of benzimidazolediones as DT-diaphorase substrates. J. Med. Chem. 2002, 45, 1211–1220. [Google Scholar] [CrossRef]
- Fagan, V.; Bonham, S.; Carty, M.P.; Aldabbagh, F. One-pot double intramolecular homolytic aromatic substitution routes to dialicyclic ring fused imidazobenzimidazolequinones and preliminary analysis of anticancer activity. Org. Biomol. Chem. 2010, 8, 3149–3156. [Google Scholar] [CrossRef] [PubMed]
- Fagan, V.; Bonham, S.; McArdle, P.; Carty, M.P.; Aldabbagh, F. Synthesis and toxicity of new ring-fused imidazo[5,4-f]benzimidazolequinones and mechanism using amine N-oxide cyclizations. Eur. J. Org. Chem. 2012, 1967–1975. [Google Scholar] [CrossRef]
- Fagan, V.; Bonham, S.; Carty, M.P.; Saenz-Méndez, P.; Eriksson, L.A.; Aldabbagh, F. COMPARE analysis of the toxicity of an iminoquinone derivative of the imidazo[5,4-f]benzimidazoles with NAD(P)H:quinone oxidoreductase 1 (NQO1) activity and computational docking of quinones as NQO1 substrates. Bioorg. Med. Chem. 2012, 20, 3223–3232. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.; Coyle, R.; Kavanagh, P.; Berezin, A.A.; Lo Re, D.; Zissimou, G.A.; Koutentis, P.A.; Carty, M.P.; Aldabbagh, F. Discovery of anti-cancer activity for benzo[1,2,4]triazin-7-ones: Very strong correlation to pleurotin and thioredoxin reductase inhibition. Bioorg. Med. Chem. 2016, 24, 3565–3570. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conboy, D.; Aldabbagh, F. 6-Imino-1,2,3,4,8,9,10,11-octahydropyrido[1,2-a]pyrido[1′,2′:1,2]imidazo[4,5-f]benzimidazole-13-one: Synthesis and Cytotoxicity Evaluation. Molbank 2020, 2020, M1118. https://doi.org/10.3390/M1118
Conboy D, Aldabbagh F. 6-Imino-1,2,3,4,8,9,10,11-octahydropyrido[1,2-a]pyrido[1′,2′:1,2]imidazo[4,5-f]benzimidazole-13-one: Synthesis and Cytotoxicity Evaluation. Molbank. 2020; 2020(1):M1118. https://doi.org/10.3390/M1118
Chicago/Turabian StyleConboy, Darren, and Fawaz Aldabbagh. 2020. "6-Imino-1,2,3,4,8,9,10,11-octahydropyrido[1,2-a]pyrido[1′,2′:1,2]imidazo[4,5-f]benzimidazole-13-one: Synthesis and Cytotoxicity Evaluation" Molbank 2020, no. 1: M1118. https://doi.org/10.3390/M1118
APA StyleConboy, D., & Aldabbagh, F. (2020). 6-Imino-1,2,3,4,8,9,10,11-octahydropyrido[1,2-a]pyrido[1′,2′:1,2]imidazo[4,5-f]benzimidazole-13-one: Synthesis and Cytotoxicity Evaluation. Molbank, 2020(1), M1118. https://doi.org/10.3390/M1118