Diethyl pyrrole-2,5-dicarboxylate



Abstract
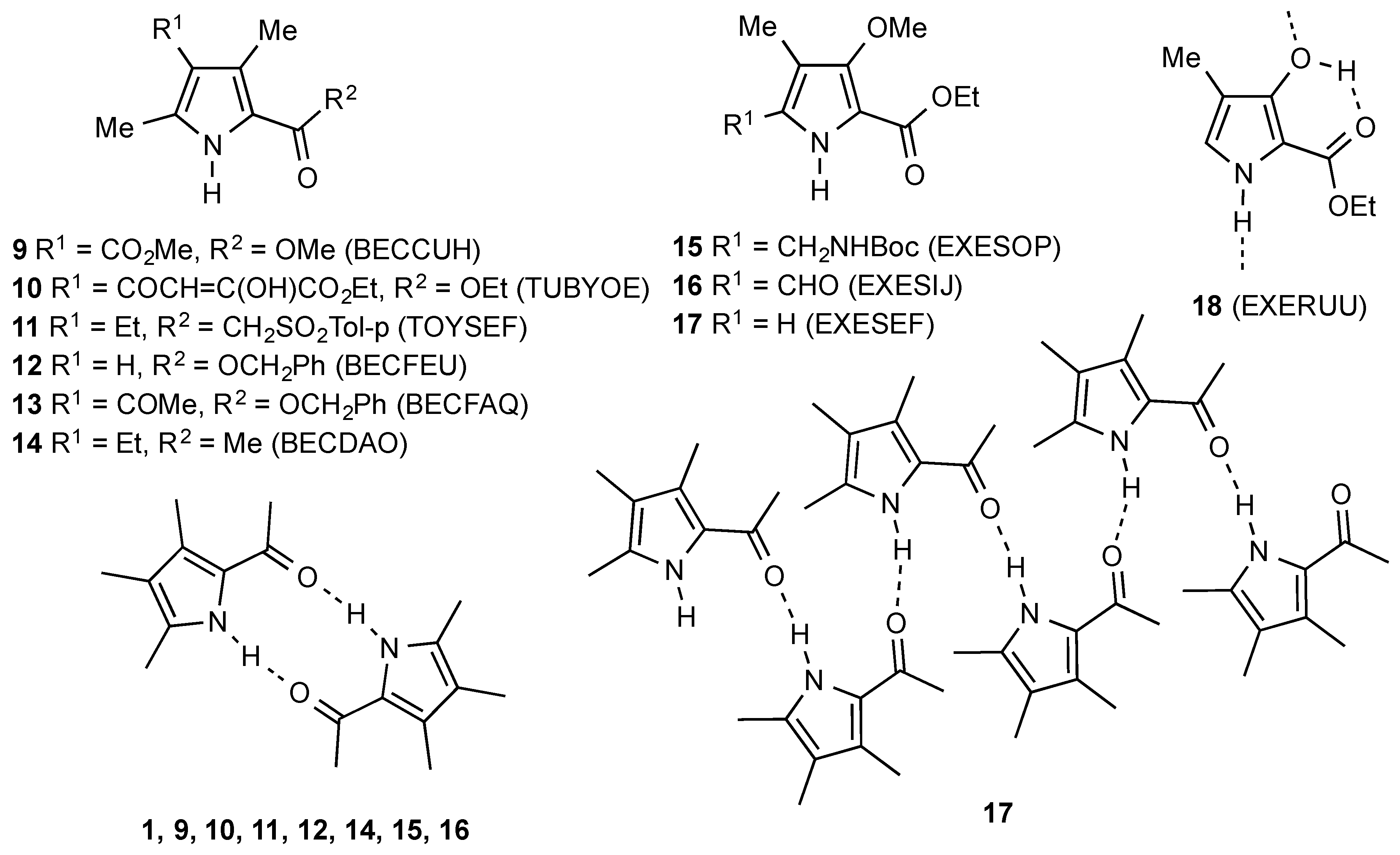
1. Introduction
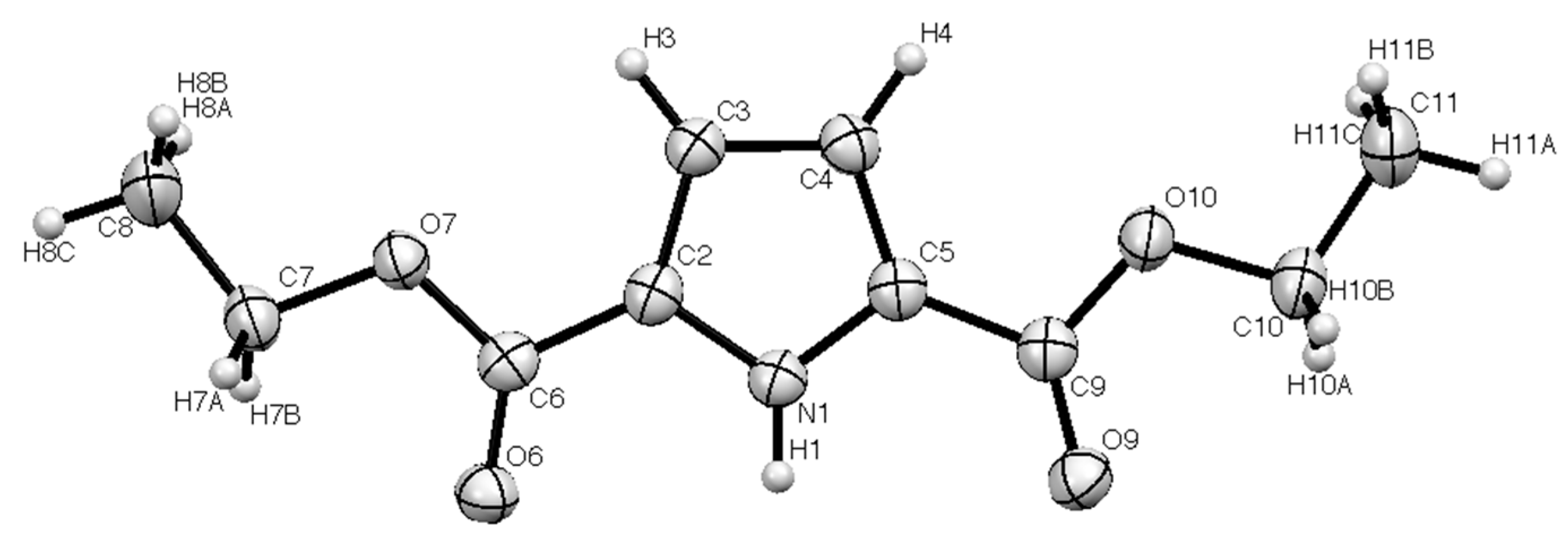
2. Results
3. Experimental
Diethyl pyrrole-2,5-dicarboxylate (1)
Supplementary Materials
Author Contributions
Conflicts of Interest
References
- Ciamician, G.; Silber, P. Ueber einige disubstituierte Derivate des Pyrrols und über ihre Constitution. Ber. Dtsch. Chem. Ges. 1886, 19, 1956–1964. [Google Scholar] [CrossRef]
- Kuhn, R.; Dury, K. Ringschlüsse mit α,α’-Dioxymuconsäure-estern. Liebigs Ann. Chem. 1951, 571, 44–68. [Google Scholar] [CrossRef]
- Ciez, D. A Direct Preparation of N-Unsubstituted Pyrrole-2,5-dicarboxylates from 2-Azidocarboxylic Esters. Org. Lett. 2009, 11, 4282–4285. [Google Scholar] [CrossRef] [PubMed]
- Salvati, M.E.; Illig, C.R.; Wilson, K.J.; Chen, J.; Meegalla, S.K.; Wall, M.J. Pyrrolopyridazine Compounds and Methods of Use Thereof for the Treatment of Proliferative Disorders. US Patent 2004 0209886 A1, 21 October 2004. [Google Scholar]
- Babu, Y.; Kotian, P.L.; Kumar, V.S.; Wu, M.; Lin, T.-H. Pyrrolo[1,2-b]pyridazine Derivatives as Janus Kinase Inhibitors. PCT International Patent Application WO 2011 014817 A1, 3 February 2011. [Google Scholar]
- Maskill, K.G.; Knowles, J.P.; Elliott, L.D.; Alder, R.W.; Booker-Milburn, K.I. Complexity from Simplicity: Tricyclic Aziridines from the Rearrangement of Pyrroles by Batch and Flow Photochemistry. Angew. Chem. Int. Ed. 2013, 52, 1499–1502. [Google Scholar] [CrossRef] [PubMed]
- Barkenbus, C.; Landis, P.S. 1,4-Thiazine. J. Am. Chem. Soc. 1948, 70, 684–685. [Google Scholar] [CrossRef]
- Berges, D.A.; Taggart, J.J. A Spontaneous Sulfoxide Dehydration. J. Org. Chem. 1985, 50, 413–415. [Google Scholar] [CrossRef]
- Marcos, C.F.; Polo, C.; Rakitin, O.A.; Rees, C.W.; Torroba, T. One-pot synthesis and chemistry of bis[1,2]dithiolopyrroles. Chem. Commun. 1997, 33, 879–880. [Google Scholar] [CrossRef]
- Rees, C.W.; White, A.J.P.; Williams, D.J.; Rakitin, O.A.; Marcos, C.F.; Polo, C.; Torroba, T. Selective Syntheses of Bis[1,2]dithiolo[1,4]thiazines and Bis[1,2]dithiolopyrroles from Hünig’s Base. J. Org. Chem. 1998, 63, 2189–2196. [Google Scholar] [CrossRef]
- Zissimou, G.A.; Kourtellaris, A.; Manoli, M.; Koutentis, P.A. Redox Active Quinoidal 1,2,4-Benzotriazines. J. Org. Chem. 2018, 83, 9391–9402. [Google Scholar] [CrossRef] [PubMed]
- Aitken, R.A.; Aitken, K.M. 1,4-Thiazines and their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry III; Katritzky, A., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; Volume 8, Chapter 9; pp. 607–675. ISBN 978-0-0804-4991-3. [Google Scholar]
- Lee, L.F.; Howe, R.K. Synthesis of 2H-1,4-Thiazine-2,6-dicarboxylates and Their Conversion to 3,4-Pyrroledicarboxylates via Sulfur Extrusion. J. Org. Chem. 1984, 49, 4780–4783. [Google Scholar] [CrossRef]
- Lee, L.F.; Schleppnik, F.M.; Howe, R.K. Syntheses and reactions of 2-halo-5-thiazolecarboxylates. J. Heterocycl. Chem. 1985, 22, 1621–1630. [Google Scholar] [CrossRef]
- Senge, M.O.; Smith, K.M. Hydrogen-bonding patterns in six derivatives of 2,4-dimethylpyrrole. Acta Crystallogr. Sect. C 2005, 61, o537–o541. [Google Scholar] [CrossRef] [PubMed]
- Bonauer, C.; Zabel, M.; König, B. Synthesis and Peptide Binding Properties of Methoxypyrrole Amino Acids (MOPAS). Org. Lett. 2004, 6, 1349–1352. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELXL. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | Length/Å | Bond | Length/Å |
|---|---|---|---|
| N(1)–C(2) | 1.366(2) | N(1)–C(5) | 1.358(2) |
| C(2)–C(6) | 1.458(2) | C(5)–C(9) | 1.467(3) |
| C(6)–O(6) | 1.214(2) | C(9)–O(9) | 1.210(2) |
| C(6)–O(7) | 1.341(2) | C(9)–O(10) | 1.348(2) |
| Angle | Value/° | Angle | Value/° |
|---|---|---|---|
| N(1)–C(2)–C(6) | 120.57(13) | N(1)–C(5)–C(9) | 121.45(13) |
| C(2)–C(6)–O(6) | 124.31(15) | C(5)–C(9)–O(9) | 125.91(16) |
| N(1)–C(2)–C(3) | 108.43(15) | N(1)–C(5)–C(4) | 108.51(16) |
| C(2)–C(3)–C(4) | 106.96(15) | C(5)–C(4)–C(3) | 107.25(14) |
| D—H…A | D—H | H…A | D…A | D—H…A |
|---|---|---|---|---|
| N(1)–H(1)…O(6) | 0.89(2) | 1.97(2) | 2.850(2) | 171(2) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aitken, R.A.; Bloomfield, C.; McGeachie, L.J.R.; Slawin, A.M.Z. Diethyl pyrrole-2,5-dicarboxylate. Molbank 2020, 2020, M1117. https://doi.org/10.3390/M1117
Aitken RA, Bloomfield C, McGeachie LJR, Slawin AMZ. Diethyl pyrrole-2,5-dicarboxylate. Molbank. 2020; 2020(1):M1117. https://doi.org/10.3390/M1117
Chicago/Turabian StyleAitken, R. Alan, Charles Bloomfield, Liam J. R. McGeachie, and Alexandra M. Z. Slawin. 2020. "Diethyl pyrrole-2,5-dicarboxylate" Molbank 2020, no. 1: M1117. https://doi.org/10.3390/M1117
APA StyleAitken, R. A., Bloomfield, C., McGeachie, L. J. R., & Slawin, A. M. Z. (2020). Diethyl pyrrole-2,5-dicarboxylate. Molbank, 2020(1), M1117. https://doi.org/10.3390/M1117