Synthesis and Isolation of Diastereomeric Anomeric Sulfoxides from a d-Mannuronate Thioglycoside Building Block


Abstract
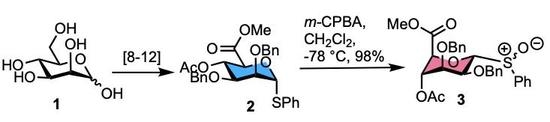
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
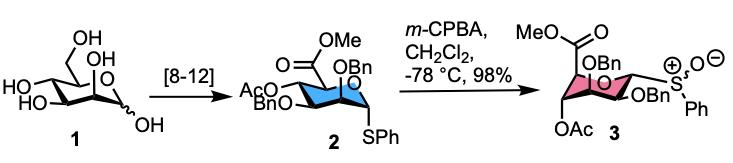
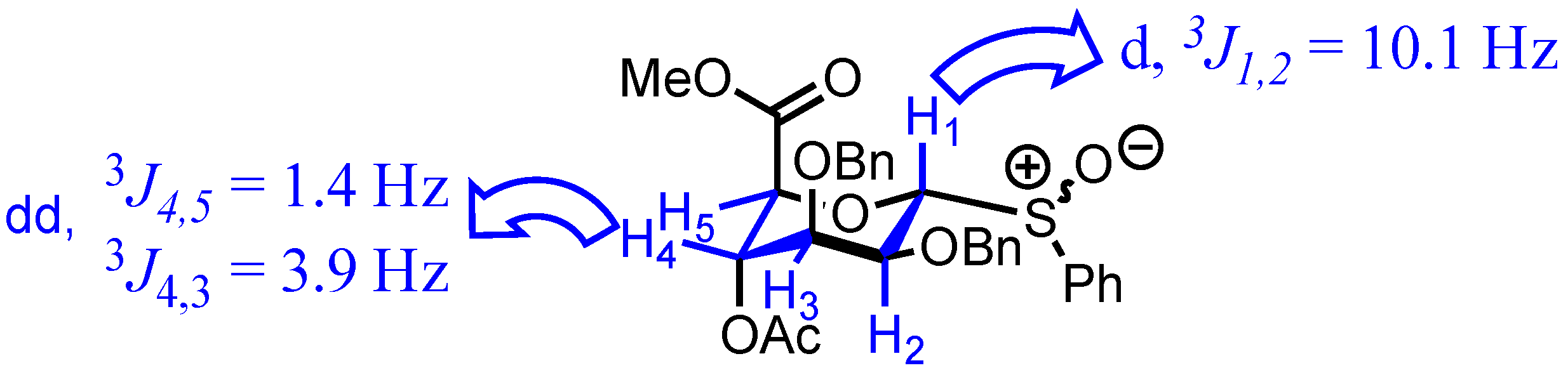
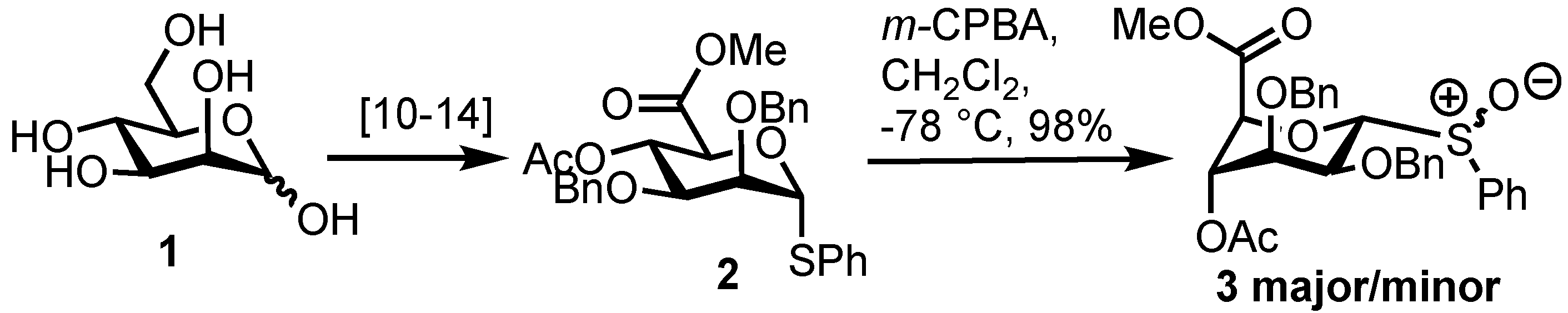
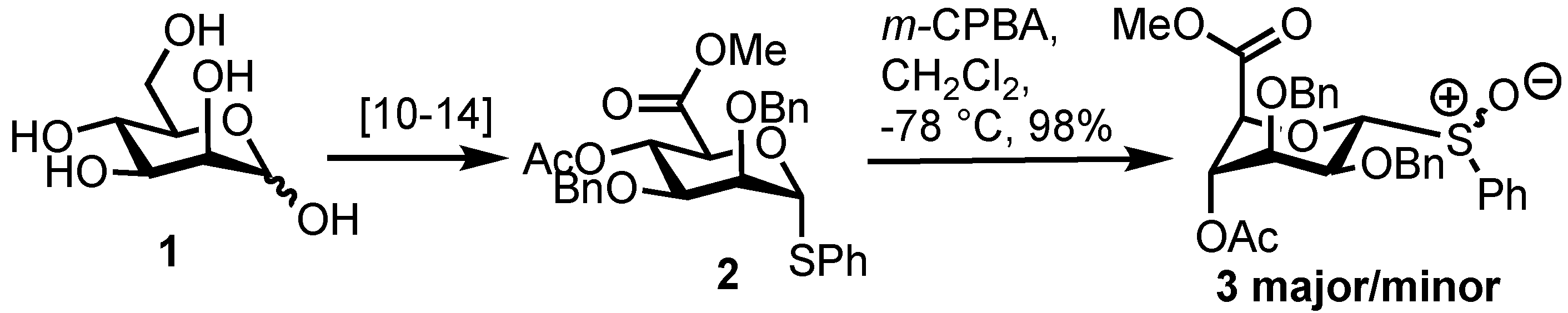
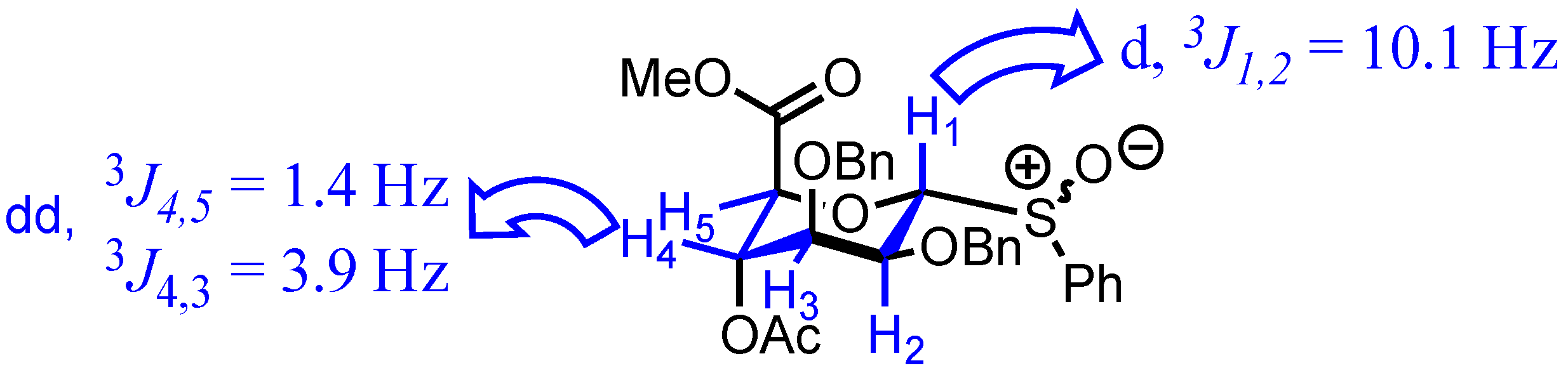
2. Results
3. Materials and Methods
3.1. General
3.2. Methyl [S-phenyl 4-O-acetyl-2,3-di-O-benzyl-1-thio-α-d-mannopyranoside (R/S)S-oxide] uronate 3
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kahne, D.; Walker, S.; Cheng, Y.; Vanengen, D.J. Glycosylation of unreactive substrates. J. Am. Chem. Soc. 1989, 111, 6881–6882. [Google Scholar] [CrossRef]
- Fascione, M.A.; Brabham, R.; Turnbull, W.B. Mechanistic Investigations into the Application of Sulfoxides in Carbohydrate Synthesis. Chem. Eur. J. 2016, 22, 3916–3928. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-Y.; Patkar, L.N.; Lin, C.-C. Selective Oxidation of Glycosyl Sulfides to Sulfoxides Using Magnesium Monoperoxyphthalate and Microwave Irradiation. J. Org. Chem. 2004, 69, 2884–2887. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-Y.; Patkar, L.N.; Chen, H.-T.; Lin, C.-C. An efficient and selective method for preparing glycosyl sulfoxides by oxidizing glycosyl sulfides with OXONE or t-BuOOH on SiO2. Carbohydr. Res. 2003, 338, 1327–1332. [Google Scholar] [CrossRef]
- Crich, D.; Mataka, J.; Zakharov, L.N.; Rheingold, A.L.; Wink, D.J.J. Stereoselective Formation of Glycosyl Sulfoxides and Their Subsequent Equilibration: Ring Inversion of an α-Xylopyranosyl Sulfoxide Dependent on the Configuration at Sulfur. J. Am. Chem. Soc. 2002, 124, 6028–6036. [Google Scholar] [CrossRef] [PubMed]
- Crich, D.; Mataka, J.; Sun, S.; Wink, D.J.; Lam, K.C.; Rheingold, A.L. Stereoselective sulfoxidation of α-mannopyranosyl thioglycosides: The exo-anomeric effect in action. Chem. Commun. 1998, 2763–2764. [Google Scholar] [CrossRef]
- Poláková, M.; Jankovič, Ľ.; Kucková, L.; Kožíšek, J. Application of oxone immobilized on montmorillonite for an efficient oxidation of mannose thioglycoside. Monatsh. Chem. 2013, 144, 969–973. [Google Scholar] [CrossRef]
- Bindschädler, P.; Noti, C.; Castagnetti, E.; Seeberger, P.H. Synthesis of a Potential 10E4 Tetrasaccharide Antigen Involved in Scrapie Pathogenesis. Helv. Chim. Acta 2006, 89, 2591–2610. [Google Scholar] [CrossRef]
- Dimitriou, E.; Miller, G.J. Exploring a glycosylation methodology for the synthesis of hydroxamate-modified alginate building blocks. Org. Biomol. Chem. 2019, 17, 9321–9335. [Google Scholar] [CrossRef] [PubMed]
- Watt, J.A.; Williams, S.J. Rapid, iterative assembly of octyl α-1,6-oligomannosides and their 6-deoxy equivalents. Org. Biomol. Chem. 2005, 3, 1982–1992. [Google Scholar] [CrossRef] [PubMed]
- Van der Marel, G.; Codée, J.D.C. Carbohydrate Chemistry: Proven Synthetic Methods; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2014; Volume 2, pp. 175–182. [Google Scholar]
- Oshitari, T.; Shibasaki, M.; Yoshizawa, T.; Tomita, M.; Takao, K.I.; Kobayashi, S. Synthesis of 2-O-(3-O-carbamoyl-α-d-mannopyranosyl)-l-gulopyranose: Sugar moiety of antitumor antibiotic bleomycin. Tetrahedron 1997, 53, 10993–11006. [Google Scholar] [CrossRef]
- Crich, D.; Smith, M.J. Solid-Phase Synthesis of β-Mannosides. J. Am. Chem. Soc. 2002, 124, 8867–8869. [Google Scholar] [CrossRef] [PubMed]
- Van den Bos, E.J.L.; Dinkelaar, J.; Overkleeft, H.S.; van der Marel, G.A.J. Stereocontrolled Synthesis of β-d-Mannuronic Acid Esters: Synthesis of an Alginate Trisaccharide. J. Am. Chem. Soc. 2006, 128, 13066–13067. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dimitriou, E.; Miller, G.J. Synthesis and Isolation of Diastereomeric Anomeric Sulfoxides from a d-Mannuronate Thioglycoside Building Block. Molbank 2020, 2020, M1111. https://doi.org/10.3390/M1111
Dimitriou E, Miller GJ. Synthesis and Isolation of Diastereomeric Anomeric Sulfoxides from a d-Mannuronate Thioglycoside Building Block. Molbank. 2020; 2020(1):M1111. https://doi.org/10.3390/M1111
Chicago/Turabian StyleDimitriou, Eleni, and Gavin J. Miller. 2020. "Synthesis and Isolation of Diastereomeric Anomeric Sulfoxides from a d-Mannuronate Thioglycoside Building Block" Molbank 2020, no. 1: M1111. https://doi.org/10.3390/M1111
APA StyleDimitriou, E., & Miller, G. J. (2020). Synthesis and Isolation of Diastereomeric Anomeric Sulfoxides from a d-Mannuronate Thioglycoside Building Block. Molbank, 2020(1), M1111. https://doi.org/10.3390/M1111