Half-Sandwich Arene-Osmium(II) Complexes with Phosphinite Ligands



Abstract
1. Introduction
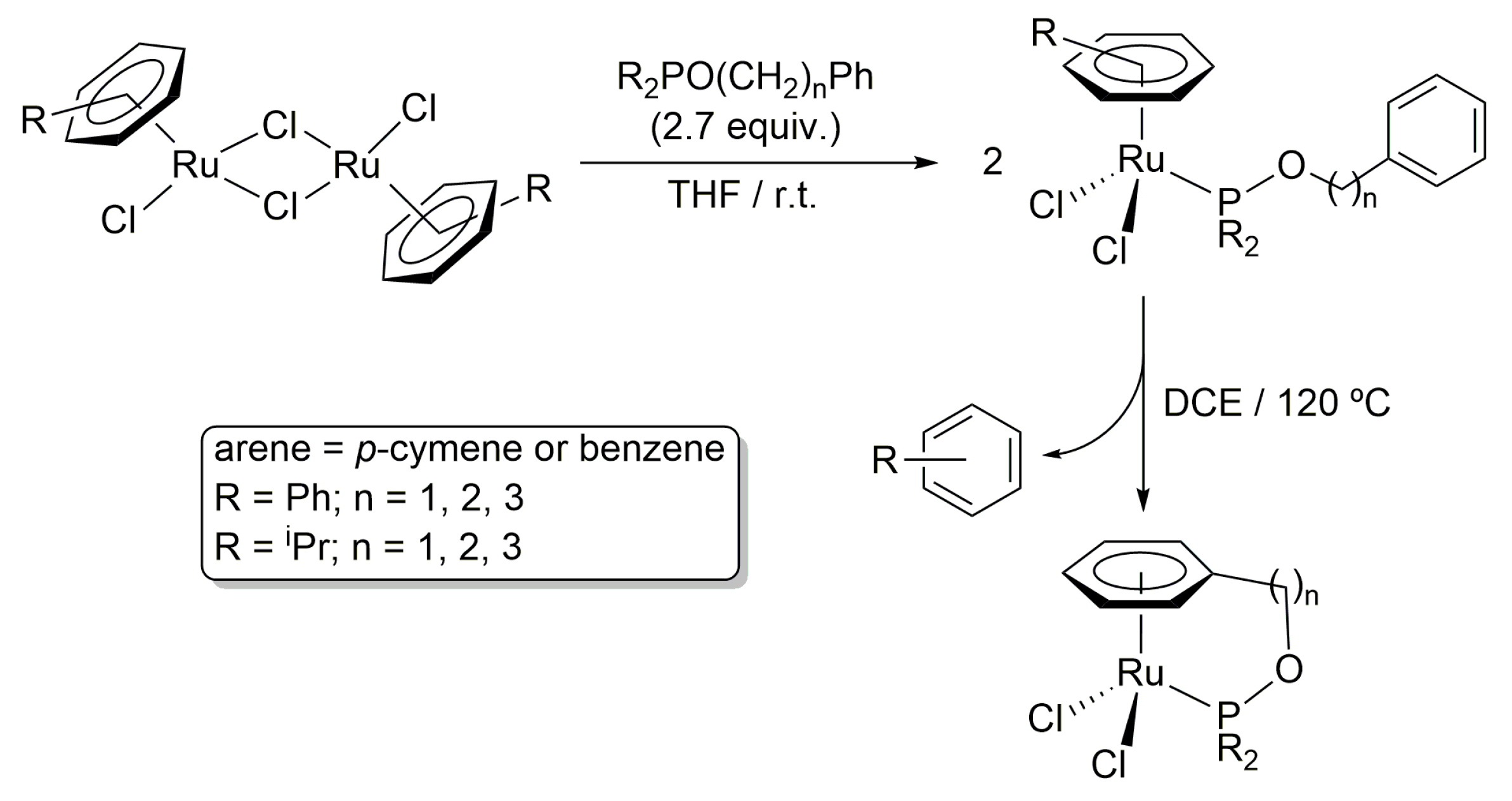
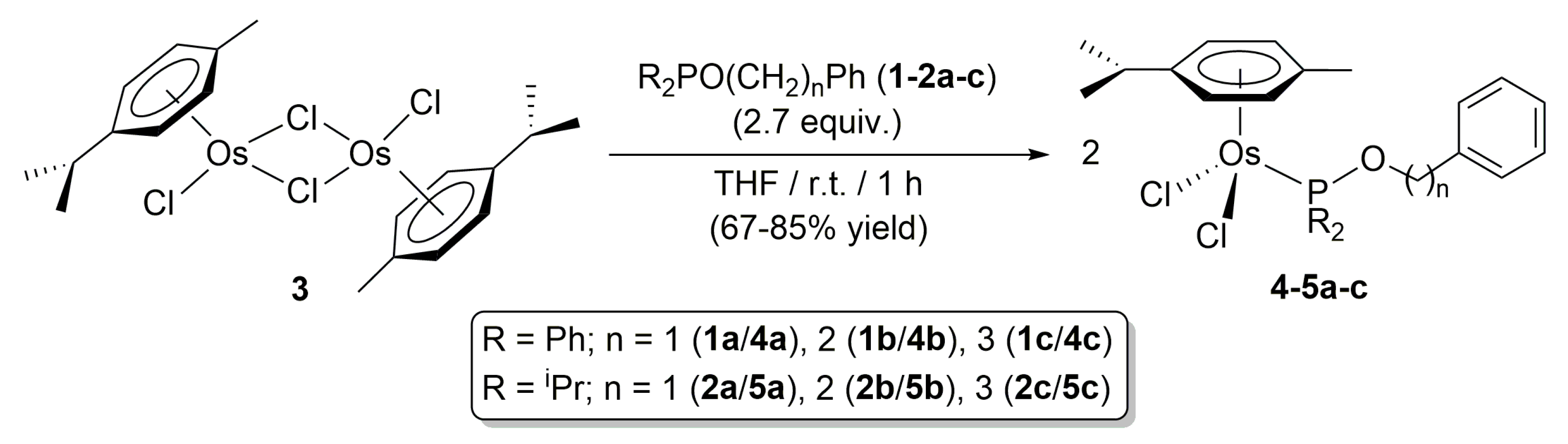
2. Results and Discussion
3. Materials and Methods
3.1. General Procedure for the Preparation of Complexes [OsCl2(η6-p-cymene){R2PO(CH2)nPh}] (R = Ph, n = 1 (4a), 2 (4b), 3 (4c); R = iPr, n = 1 (5a), 2 (5b), 3 (5c))
3.1.1. [OsCl2(η6-p-cymene)(Ph2POCH2Ph)] (4a)
3.1.2. [OsCl2(η6-p-cymene)(Ph2POCH2CH2Ph)] (4b)
3.1.3. [OsCl2(η6-p-cymene)(Ph2POCH2CH2CH2Ph)] (4c)
3.1.4. [OsCl2(η6-p-cymene)(iPr2POCH2Ph)] (5a)
3.1.5. [OsCl2(η6-p-cymene)(iPr2POCH2CH2Ph)] (5b)
3.1.6. [OsCl2(η6-p-cymene)(iPr2POCH2CH2CH2Ph)] (5c)
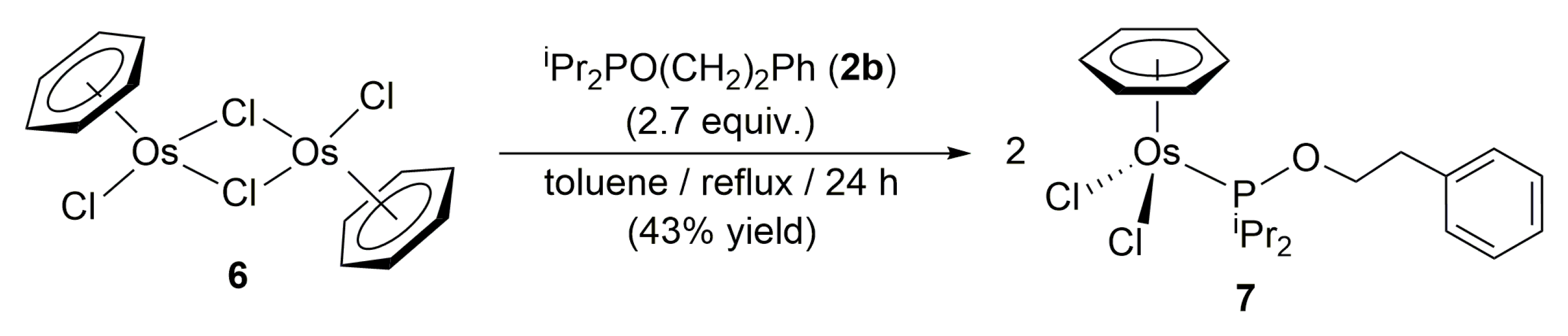
3.2. Preparation of Complex [OsCl2(η6-benzene){iPr2PO(CH2)2Ph}] (7)
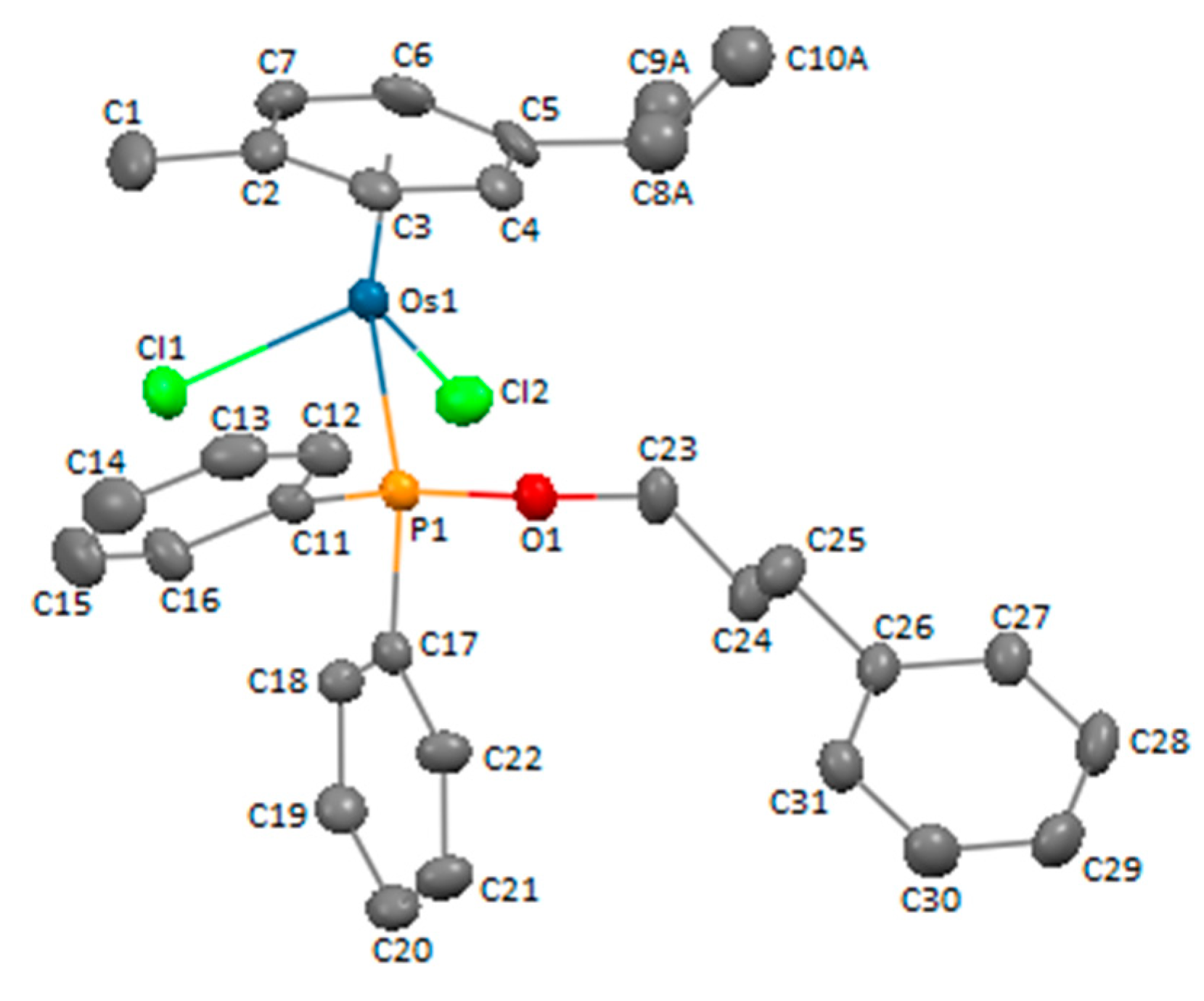
3.3. X-ray Crystal Structure Determination of Compound 4c
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- González-Fernández, R.; Crochet, P.; Cadierno, V. Half-sandwich ruthenium(II) complexes with tethered arene-phosphinite ligands: Synthesis, structure and application in catalytic cross dehydrogenative coupling reactions of silanes and alcohols. Dalton Trans. 2020, 49, 210–222. [Google Scholar] [CrossRef]
- Miyaki, Y.; Onishi, T.; Kurosawa, H. Synthesis and reaction of ruthenium(II) complexes containing heteroatom donor (O, N, P) tethered to η6-arene ring. Inorg. Chim. Acta 2000, 300–302, 369–377. [Google Scholar] [CrossRef]
- Weber, I.; Heinemann, F.W.; Bauer, W.; Superchi, S.; Zahl, A.; Richter, D.; Zenneck, U. Diastereoselective routes to [Amino{σ(P):η6-(ansa-phosphinite)benzene}chlororuthenium(II)]PF6 salts: Kinetic versus thermodynamic preferences. Organometallics 2008, 27, 4116–4125. [Google Scholar] [CrossRef]
- Pandey, M.K.; Mague, J.T.; Balakrishna, M.S. Sterically demanding phosphines with 2,6-dibenzhydryl-4-methylphenyl core: Synthesis of RuII, PdII, and PtII complexes, and structural and catalytic studies. Inorg. Chem. 2018, 57, 7468–7480. [Google Scholar] [CrossRef]
- Bennett, M.A.; Harper, J.R. Tethered arene complexes of ruthenium. In Modern Coordination Chemistry; Leigh, G.J., Winterton, N., Eds.; RSC Publishing: Cambridge, UK, 2002; pp. 163–168. [Google Scholar]
- Clark, T.J.; Lee, K.; Manners, I. Transition-metal-catalyzed dehydrocoupling: A convenient route to bonds between main-group elements. Chem. Eur. J. 2006, 12, 8634–8648. [Google Scholar] [CrossRef]
- Kuciński, K.; Hreczycho, G. Catalytic formation of silicon-heteroatom (N, P, O, S) bonds. ChemCatChem 2017, 9, 1868–1885. [Google Scholar] [CrossRef]
- Ventura-Espinosa, D.; Sabater, S.; Carretero-Cerdán, A.; Baya, M.; Mata, J.A. High production of hydrogen on demand from silanes catalyzed by iridium complexes as a versatile hydrogen storage system. ACS Catal. 2018, 8, 2558–2566. [Google Scholar] [CrossRef]
- Esteruelas, M.A.; López, A.M.; Oñate, E.; Royo, E. Synthesis and reactivity of osmium complexes containing a cyclopentadienyl ligand with a pendant phosphine donor group. Organometallics 2004, 23, 3021–3030. [Google Scholar] [CrossRef]
- Batuecas, M.; Esteruelas, M.A.; García-Yebra, C.; Oñate, E. Redox isomerization of allylic alcohols catalyzed by osmium and ruthenium complexes containing a cyclopentadienyl ligand with a pendant amine or phosphoramidite group: X-ray structure of an η3-1-hydroxyallyl-metal-hydride intermediate. Organometallics 2010, 29, 2166–2175. [Google Scholar] [CrossRef]
- Baya, M.; Crochet, P.; Esteruelas, M.A.; Oñate, E. Generation of functionally substituted cyclopentadienyl ligands in osmium(IV) chemistry. Organometallics 2001, 20, 240–253. [Google Scholar] [CrossRef]
- Braunschweig, H.; Dellermann, T.; Dewhurst, R.D.; Mies, J.; Radacki, K.; Stellwag-Konertz, S.; Vargas, A. Strained ansa half-sandwich complexes of ruthenium and osmium and a non-iron metallopolymer by ring-opening polymerization. Organometallics 2014, 33, 1536–1539. [Google Scholar] [CrossRef]
- Zhou, X.; He, X.; Lin, J.; Zhuo, Q.; Chen, Z.; Zhang, H.; Wang, J.; Xia, H. Reactions of osmium hydrido alkenylcarbyne with allenoates: Insertion and [3 + 2] annulation. Organometallics 2015, 34, 1742–1750. [Google Scholar] [CrossRef]
- Bell, A.G.; Koźmiński, W.; Linden, A.; von Philipsborn, W. 187Os NMR study of (η6-arene)osmium(II) complexes: Separation of electronic and steric ligand effects. Organometallics 1996, 15, 3124–3135. [Google Scholar] [CrossRef]
- González-Fernández, R.; Crochet, P.; Cadierno, V.; Menéndez, M.I.; López, R. Phosphinous acid-assisted hydration of nitriles: Understanding the controversial reactivity of osmium and ruthenium catalysts. Chem. Eur. J. 2017, 23, 15210–15221. [Google Scholar] [CrossRef]
- Freedman, D.A.; Magneson, D.J.; Mann, K.R. Synthesis, characterization, and photochemistry of bis(arene) and bis(acetonitrile) complexes of osmium(II). Inorg. Chem. 1995, 34, 2617–2624. [Google Scholar] [CrossRef]
- Freedman, D.A.; Gill, T.P.; Blough, A.M.; Koefod, R.S.; Mann, K.R. Preparation and reactions of [CpOs(CH3CN)3]+. A useful synthetic intermediate for the preparation of CpOsL3 compounds. Inorg. Chem. 1997, 36, 95–102. [Google Scholar] [CrossRef]
- Wang, R.; Eberspacher, T.A.; Hasegawa, T.; Day, V.; Ware, D.C.; Taube, H. Tmtacn, tacn, and triammine complexes of (η6-arene)OsII: Syntheses, characterizations, and photosubstitution reactions (tmtacn = 1,4,7-trimethyl-1,4,7-triazacyclononane; tacn = 1,4,7-triazacyclononane). Inorg. Chem. 2001, 40, 593–600. [Google Scholar] [CrossRef]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 5th ed.; Butterworth-Heinemann: Oxford, UK, 2003. [Google Scholar]
- Masutani, K.; Minowa, T.; Hagiwara, Y.; Mukaiyama, T. Cyanation of alcohols with diethyl cyanophosphonate and 2,6-dimethyl-1,4-benzoquinone by a new type of oxidation-reduction condensation. Bull. Chem. Soc. Jpn. 2006, 79, 1106–1117. [Google Scholar] [CrossRef]
- Shintou, T.; Kikuchi, W.; Mukaiyama, T. Efficient method for the preparation of carboxylic acid alkyl esters or alkyl phenyl ethers by a new type of oxidation-reduction condensation using 2,6-dimethyl-1,4-benzoquinone and alkoxydiphenylphosphines. Bull. Chem. Soc. Jpn. 2003, 76, 1645–1667. [Google Scholar] [CrossRef]
- Castarlenas, R.; Esteruelas, M.A.; Oñate, E. N-Heterocyclic carbene-osmium complexes for olefin metathesis reactions. Organometallics 2005, 24, 4343–4346. [Google Scholar] [CrossRef]
- Arthur, T.; Stephenson, T.A. Synthesis of triple halide-bridged arene complexes of ruthenium(II) and osmium(II). J. Organomet. Chem. 1981, 208, 369–387. [Google Scholar] [CrossRef]
- CrysAlisPro CCD & CrysAlisPro RED; Oxford Diffraction Ltd.: Oxford, UK, 2008.
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Beurskens, P.T.; Beurskens, G.; de Gelder, R.; García-Granda, S.; Gould, R.O.; Smits, J.M.M. DIRDIF2008 Program System. Crystallography Laboratory; University of Nijmegen: Nijmegen, The Netherlands, 2008. [Google Scholar]
- Sheldrick, G.M. SHELXL97: Program for the Refinement of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Wilson, A.J.C. (Ed.) International Tables for X-ray Crystallography, Volume, C; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1992. [Google Scholar]
- Nardelli, M. PARST: A system of FORTRAN routines for calculating molecular structure parameters from results of crystal structure analyses. Comput. Chem. 1983, 7, 95–98. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | δP (ppm) | Complex | δP (ppm) |
|---|---|---|---|
| 1a | 114.5 | 4a | 64.2 |
| 1b | 126.6 | 4b | 63.1 |
| 1c | 112.1 | 4c2 | 62.2 |
| 2a | 154.5 | 5a2 | 103.5 |
| 2b | 152.3 | 5b2 | 103.3 |
| 2c | 151.4 | 5c | 102.3 |
| Chemical Formula | C31H35Cl2OOsP |
|---|---|
| fw | 715.66 |
| T (K) | 293(2) |
| cryst. syst. | monoclinic |
| space group | P 21/c |
| cryst. size mm3 | 0.24 × 0.16 × 0.16 |
| a, Å | 10.3465(2) |
| b, Å | 19.1010(3) |
| c, Å | 15.1007(3) |
| α, deg | 90 |
| β, deg | 105.009(2) |
| γ, deg | 90 |
| Z | 4 |
| V, Å3 | 2882.5(1) |
| ρcalcd, g cm–3 | 1.649 |
| μ, mm−1 | 10.751 |
| F(000) | 1416 |
| θ range, deg | 3.813 to 69.621 |
| index ranges | −12 ≤ h ≤ 12; −22 ≤ k ≤ 22; −18 ≤ l ≤ 13 |
| completeness to θmax | 97.9% |
| no. of data collected | 14414 |
| no. of unique data | 5321 |
| no. of parameters/restrains | 319/4 |
| refinement method | full-matrix least-squares on F2 |
| goodness of fit on F2 | 1.065 |
| R1a [I > 2σ(I)] | 0.0407 |
| wR2a [I > 2σ(I)] | 0.0978 |
| R1 (all data) | 0.0486 |
| wR2 (all data) | 0.1033 |
| largest diff. peak and hole, e Å−3 | 0.935 and −1.683 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Fernández, R.; Borge, J.; Crochet, P.; Cadierno, V. Half-Sandwich Arene-Osmium(II) Complexes with Phosphinite Ligands. Molbank 2020, 2020, M1110. https://doi.org/10.3390/M1110
González-Fernández R, Borge J, Crochet P, Cadierno V. Half-Sandwich Arene-Osmium(II) Complexes with Phosphinite Ligands. Molbank. 2020; 2020(1):M1110. https://doi.org/10.3390/M1110
Chicago/Turabian StyleGonzález-Fernández, Rebeca, Javier Borge, Pascale Crochet, and Victorio Cadierno. 2020. "Half-Sandwich Arene-Osmium(II) Complexes with Phosphinite Ligands" Molbank 2020, no. 1: M1110. https://doi.org/10.3390/M1110
APA StyleGonzález-Fernández, R., Borge, J., Crochet, P., & Cadierno, V. (2020). Half-Sandwich Arene-Osmium(II) Complexes with Phosphinite Ligands. Molbank, 2020(1), M1110. https://doi.org/10.3390/M1110