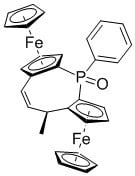
(2Sp,4R,8Sp)-4-Methyl-1-phenyl-diferroceno-5-Z-ethylene-1-phosphinoxide



Abstract
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Synthesis
3.2.1. 1,1-(Phenylphosphinidene)di[(2Sp)-2-vinyl]ferrocene 2:
3.2.2. (2Sp,4R,8Sp)-4-Methyl-1-phenyl-diferroceno-5-Z-ethylene-1-phosphinoxide 3:
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schaarschmidt, D.; Lang, H. Selective Syntheses of Planar-Chiral Ferrocenes. Organometallics 2013, 32, 5668–5704. [Google Scholar] [CrossRef]
- Štěpnička, P. Ferrocenes: Ligands, Materials and Biomolecules; John Wiley & Son: Chichester, UK, 2008. [Google Scholar]
- Blaser, H.-U.; Pugin, B.; Spindler, F.; Thommen, M. From a chiral switch to a ligand portfolio for asymmetric catalysis. Acc. Chem. Res. 2007, 40, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Barbaro, P.; Bianchini, C.; Togni, A. Synthesis and Characterization of Ruthenium (II) Complexes Containing Chiral Bis (ferrocenyl)−P3 or −P2S Ligands. Asymmetric Transfer Hydrogenation of Acetophenone. Organometallics 1997, 16, 3004–3014. [Google Scholar] [CrossRef]
- Xiao, L.; Weissensteiner, W.; Widhalm, M. Novel chiral biferrocene ligands for palladium-catalyzed allylic substitution reactions. J. Org. Chem. 2002, 67, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, L.; Lampin, J.P.; Mathey, F. Untersuchung der Metallierung von Diferrocenylphenylphosphinoxid. J. Organomet. Chem. 1974, 80, 109–118. [Google Scholar] [CrossRef]
- Metallinos, C.; Szillat, H.; Taylor, N.; Snieckus, V. (−)-Sparteine-Mediated Directed ortho-Metalation of N-Cumyl-N-ethylferrocenecarboxamide. Versatile Routes to Functionalized Planar Chiral Ferrocenecarboxamides, Amines, Esters and Phosphines. Adv. Synth. Catal. 2003, 345, 370–382. [Google Scholar] [CrossRef]
- Tews, D.; Gaede, P. Syntheses of Dinuclear Metal Complexes of Rhodium, Iridium, Iron, Molybdenum, and Cobalt with Novel Bridged 2,2’-Bis (indenyl) Systems. Organometallics 2004, 23, 968–975. [Google Scholar] [CrossRef]
- Santi, S.; Orian, L.; Durante, C.; Bencze, E.; Bisello, A.; Donoli, A.; Ceccon, A.; Benetollo, F.; Crociani, L. Metal–Metal Electronic Coupling in syn and anti Stereoisomers of Mixed-Valent (FeCp)2-, (RhL2)2-, and (FeCp)(RhL2)-as-Indacenediide Ions. Chem. Eur. J. 2007, 13, 7933–7947. [Google Scholar] [CrossRef]
- Chen, J.; Murillo Parra, D.; Lalancette, R.; Jäkle, F. Redox-Switchable Chiral Anions and Cations Based on Heteroatom-Fused Biferrocenes. Organometallics 2015, 34, 4323–4330. [Google Scholar] [CrossRef]
- Köhler, F.; Schell, A.; Weber, B. Tuning the link of doubly silyl-bridged ferrocenes. J. Organomet. Chem. 1999, 575, 33–38. [Google Scholar] [CrossRef]
- Nagahora, N.; Ogawa, S.; Kawai, Y.; Sato, R. First synthesis and structure of sulfur-containing heterocycles fused to ferrocene. Tetrahedron Lett. 2002, 43, 5825–5828. [Google Scholar] [CrossRef]
- Venkatasubbaiah, K.; Zakharov, L.; Kassel, W.; Rheingold, A.; Jäkle, F. Reversible Expansion and Contraction of a 1,2-Diborylated Ferrocene Dimer Promoted by Redox Chemistry and Nucleophile Binding. Angew. Chem. Int. Ed. 2005, 44, 5428–5433. [Google Scholar] [CrossRef] [PubMed]
- Venkatasubbaiah, K.; Pakkirisamy, T.; Lalancette, R.; Jäkle, F. Tuning the electronic structure of diboradiferrocenes. Dalton Trans. 2008, 33, 4507–4513. [Google Scholar] [CrossRef] [PubMed]
- Thilagar, P.; Murillo, D.; Chen, J.; Jäkle, F. Synthesis and supramolecular assembly of the bifunctional borinic acid [1,2-fcB(OH)]2. Dalton Trans. 2013, 42, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Murillo Parra, D.; Lalancette, R.; Jäkle, F. Diferrocenophosphaborin: A Planar-Chiral, Redox-Active and Anion-Responsive Ambiphilic Ligand. Angew. Chem. Int. Ed. 2015, 54, 10202–10205. [Google Scholar] [CrossRef]
- Honegger, P.; Widhalm, M. Formaldehyde-1,1″-(phenylphosphinidenesulfide)bis[(2S)-2-[(1R)-1-(methylamino)-ethyl]]ferrocene-aminal. Molbank 2019, 4, M1090. [Google Scholar] [CrossRef]
- Honegger, P.; Widhalm, M. (2S,4R,6R,8S)-4,6-Dimethyl-1-phenyl-diferroceno-1-phosphines. Molbank 2019, 4, M1098. [Google Scholar] [CrossRef]
- Benedikt, M.; Schlögl, K. Syntheses and stereochemistry of metalloceno cycloocta-1,5-dienes: [2]Orthocyclo[2](1,2)benchrotrenophane and-ferrocenophane, [2.2](1,2)Ferrocenophane. Monatsh. Chem. 1978, 109, 805–822. [Google Scholar] [CrossRef]
- Atzkern, H.; Huber, B.; Köhler, F.; Müller, G.; Müller, R. Rigid bent bridges between cyclopentadienylmetal fragments. Iron derivatives of the 4,8-ethano-2,4,6,8-tetrahydro-s-indacene-2,6-diyl dianion. Organometallics 1991, 10, 238–244. [Google Scholar] [CrossRef]
- Barreiro, E.; Broggini, D.; Adrio, L.; White, A.; Schwenk, R.; Togni, A.; Hii, K. Gold (I) complexes of conformationally constricted chiral ferrocenyl phosphines. Organometallics 2012, 31, 3745–3754. [Google Scholar] [CrossRef]
- Smith, A.M.R.; Rzepa, H.S.; White, A.J.P.; Billen, D.; Hii, K.K. Delineating Origins of Stereocontrol in Asymmetric Pd-Catalyzed α-Hydroxylation of 1,3-Ketoesters. J. Org. Chem. 2010, 75, 3085–3096. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Mise, T.; Fukushima, M.; Kagotani, M.; Nagashima, N.; Hamada, Y.; Matsumoto, A.; Kawakami, S.; Konishi, M.; Yamamoto, K.; et al. Asymmetric Synthesis Catalyzed by Chiral Ferrocenylphosphine–Transition Metal Complexes. I. Preparation of Chiral Ferrocenylphosphines. Bull. Chem. Soc. Jpn. 1980, 53, 1138–1151. [Google Scholar] [CrossRef]
- Butler, I.; Cullen, W.; Rettig, S. Synthesis of derivatives of [alpha (dimethylamino)ethyl] ferrocene via lithiation reactions and the structure of 2-[alpha-(dimethylamino)ethyl]-1,1′,3-tris(trimethylsilyl)ferrocene. Organometallics 1986, 5, 1320–1328. [Google Scholar] [CrossRef]
- Gschwend, B.; Pugin, B.; Bertogg, A.; Pfaltz, A. P-Chiral Ferrocenephospholanes: Synthesis, Reactivity, Metal Complex Chemistry and Application in the Asymmetric Hydrogenation of Olefins. Chem. Eur. J. 2009, 15, 12993–13007. [Google Scholar] [CrossRef]
- Compain, P. Olefin Metathesis of Amine-Containing Systems: Beyond the Current Consensus. Adv. Synth. Catal. 2007, 349, 1829–1846. [Google Scholar] [CrossRef]
- Goldberg, S.; Loeble, W.; Tidwell, T. Alumina-catalyzed dehydration of 1-ferrocenylethanol. Formation of 1,3-diferrocenyl-1-butene. J. Org. Chem. 1967, 32, 4070–4071. [Google Scholar] [CrossRef]
- Horspool, W.; Stanley, P.; Sutherland, R.; Thomson, B. Studies on ferrocene derivatives. Part X. Reaction of acetylferrocene with methylmagnesium iodide and of ferrocenyl alcohols with Grignard reagents. J. Chem. Soc. 1971, 1365–1369. [Google Scholar] [CrossRef]
- Abram, T.; Watts, W. Stable carbocations. Part VIII. Fragmentation reactions of ferrocenylalkylium ions. J. Chem. Soc. Perkin 1975, 113–116. [Google Scholar] [CrossRef]
- Gokel, G.; Marquarding, D.; Ugi, I. Stereoselective syntheses. VIII. Retentive nucleophilic displacements of.alpha.-substituted alkylferrocenes. J. Org. Chem. 1972, 20, 3052–3058. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honegger, P.; Widhalm, M. (2Sp,4R,8Sp)-4-Methyl-1-phenyl-diferroceno-5-Z-ethylene-1-phosphinoxide. Molbank 2020, 2020, M1105. https://doi.org/10.3390/M1105
Honegger P, Widhalm M. (2Sp,4R,8Sp)-4-Methyl-1-phenyl-diferroceno-5-Z-ethylene-1-phosphinoxide. Molbank. 2020; 2020(1):M1105. https://doi.org/10.3390/M1105
Chicago/Turabian StyleHonegger, Philipp, and Michael Widhalm. 2020. "(2Sp,4R,8Sp)-4-Methyl-1-phenyl-diferroceno-5-Z-ethylene-1-phosphinoxide" Molbank 2020, no. 1: M1105. https://doi.org/10.3390/M1105
APA StyleHonegger, P., & Widhalm, M. (2020). (2Sp,4R,8Sp)-4-Methyl-1-phenyl-diferroceno-5-Z-ethylene-1-phosphinoxide. Molbank, 2020(1), M1105. https://doi.org/10.3390/M1105