Formaldehyde-1,1″-(phenylphosphinidenesulfide)bis[(2S)-2-[(1R)-1-(methylamino)-ethyl]]ferrocene-aminal



Abstract
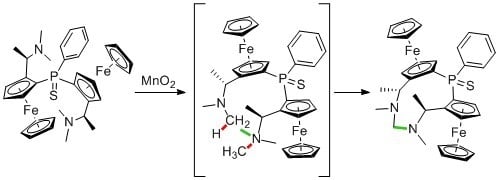
:
{kind=link}
{kind=link}
1. Introduction
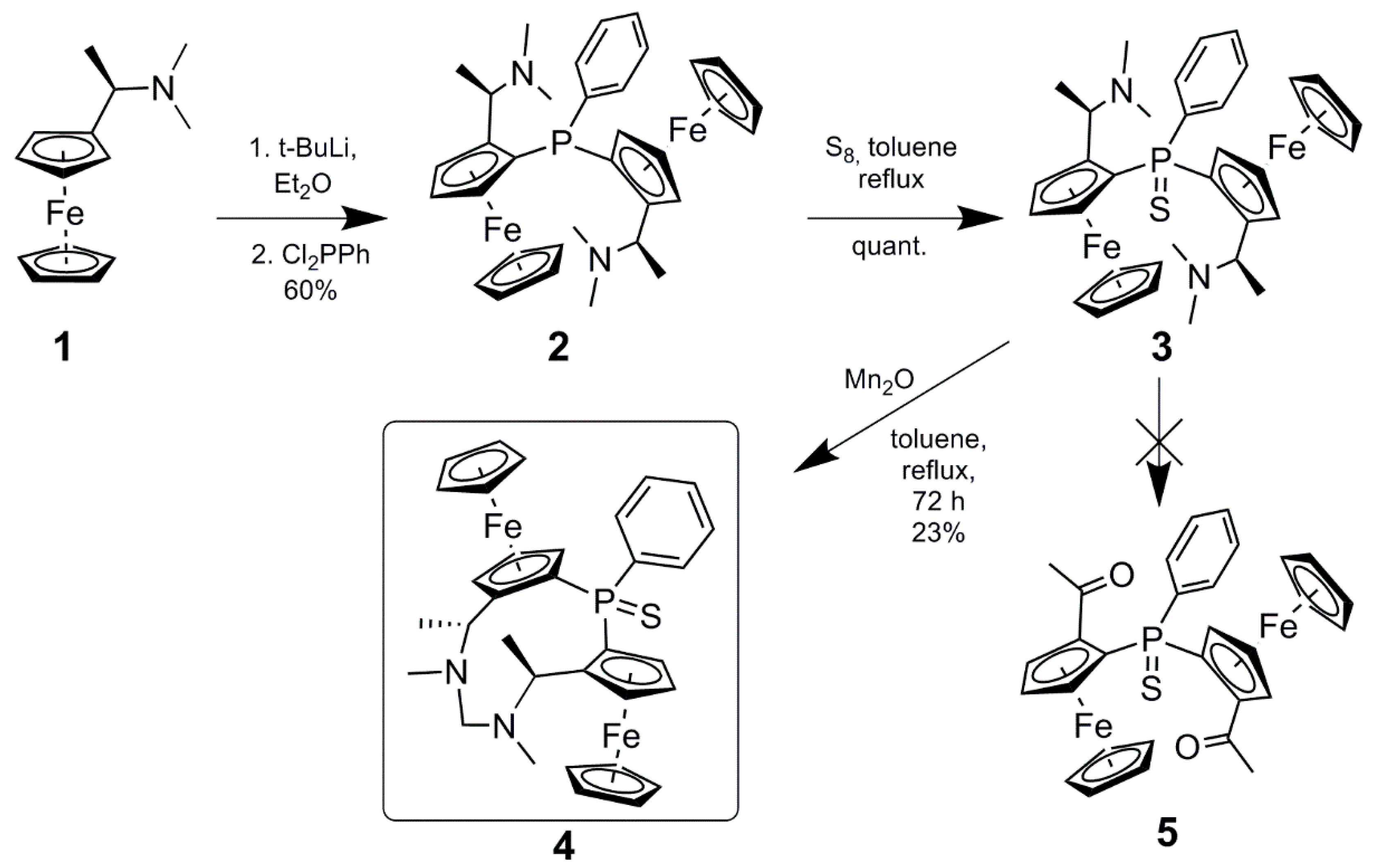
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Synthesis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Blaser, H.-U.; Pugin, B.; Spindler, F.; Thommen, M. From a chiral switch to a ligand portfolio for asymmetric catalysis. Acc. Chem. Res. 2007, 40, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Weissensteiner, W.; Widhalm, M. Novel chiral biferrocene ligands for palladium-catalyzed allylic substitution reactions. J. Org. Chem. 2002, 67, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Nishibayashi, Y.; Segawa, K.; Singh, J.; Fukuzawa, S.; Ohe, K.; Uemura, S. Novel chiral ligands, diferrocenyl dichalcogenides and their derivatives, for rhodium-and iridium-catalyzed asymmetric hydrosilylation. Organometallics 1996, 15, 370–379. [Google Scholar] [CrossRef]
- Barbaro, P.; Bianchini, C.; Togni, A. Synthesis and Characterization of Ruthenium(II) Complexes Containing Chiral Bis (ferrocenyl)-P3 or -P2S Ligands. Asymmetric Transfer Hydrogenation of Acetophenone. Organometallics 1997, 16, 3004–3014. [Google Scholar] [CrossRef]
- Barbaro, P.; Bianchini, C.; Oberhauser, W.; Togni, A. Synthesis and characterization of chiral bis-ferrocenyl triphosphine Ni(II) and Rh(III) complexes and their use as catalyst precursors for acetalization reactions. J. Mol. Catal. 1999, 145, 139–146. [Google Scholar] [CrossRef]
- Fadini, L.; Togni, A. Ni(II)-PPP complexes: From the hydroamination of activated olefins to the synthesis of β-amino acids. Chimia 2004, 58, 208–211. [Google Scholar] [CrossRef]
- Wang, Y.; Weissensteiner, W.; Mereiter, K.; Spindler, F. Preparation of C2-Symmetric Bis [2-(diphenylphosphino)ferrocen-1-yl]-methane and Its Use in Rhodium- and Ruthenium-Catalyzed Hydrogenation. Helv. Chim. Acta 2006, 89, 1772–1782. [Google Scholar] [CrossRef]
- Barreiro, E.; Broggini, D.; Adrio, L.; White, A.; Schwenk, R.; Togni, A.; Hii, K. Gold(I) complexes of conformationally constricted chiral ferrocenyl phosphines. Organometallics 2012, 31, 3745–3754. [Google Scholar] [CrossRef]
- Hu, H.; Tang, Q.; Tu, A.; Zhong, W. Chiral Bifunctional Ferrocenylphosphines as New Catalysts for aza-Morita-Baylis-Hillman Reactions of N-Sulfonated Imines with Methyl Vinyl Ketone. Curr. Organocatalysis 2015, 2, 58–63. [Google Scholar] [CrossRef]
- Fleury-Bregeot, N.; Jean, L.; Retailleau, P.; Martinetti, A. Screening of chiral phosphines as catalysts for the enantioselective [3+2] annulations of N-tosylimines with allenic esters. Tetrahedron 2007, 63, 11920–11927. [Google Scholar] [CrossRef]
- Hu, H.; Yu, S.; Zhu, L.; Zhou, L.; Zhong, W. Chiral bifunctional ferrocenylphosphine catalyzed highly enantioselective [3+2] cycloaddition reaction. Org. Biomol. Chem. 2016, 14, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Marquarding, D.; Klusacek, H.; Gokel, G.; Hoffmann, P.; Ugi, I. Stereoselective syntheses. VI. Correlation of central and planar chirality in ferrocene derivatives. J. Am. Chem. Soc. 1970, 92, 5389–5393. [Google Scholar] [CrossRef]
- Hayashi, T.; Mise, T.; Fukushima, M.; Kagotani, M.; Nagashima, N.; Hamada, Y.; Matsumoto, A.; Kawakami, S.; Konishi, M.; Yamamoto, K.; et al. Asymmetric Synthesis Catalyzed by Chiral Ferrocenylphosphine–Transition Metal Complexes. I. Preparation of Chiral Ferrocenylphosphines. Bull. Chem. Soc. Jpn. 1980, 53, 1138–1151. [Google Scholar] [CrossRef]
- Butler, I.; Cullen, W.; Rettig, S. Synthesis of derivatives of [.alpha.(dimethylamino)ethyl]ferrocene via lithiation reactions and the structure of 2-[.alpha.-(dimethylamino)ethyl]-1,1′,3-tris(trimethylsilyl)ferrocene. Organometallics 1986, 5, 1320–1328. [Google Scholar] [CrossRef]
- Fleischer, I.; Toma, Š. Synthesis of New Chiral 1,2-Disubstituted Ferrocenes. Collect. Czechoslov. Chem. Commun. 2004, 69, 330–338. [Google Scholar] [CrossRef]
- Tobrmann, T.; Dalimil, D. Regioselective and Facile Synthesis of 7,9-Dialkyl-8-oxopurines from 7,9-Dialkyl-7,8-dihydropurines: Total Synthesis of Heteromines I and J. Synthesis 2014, 46, 660–668. [Google Scholar] [CrossRef]
- Brecker, L.; Kögl, F.; Tyl, C.; Kratzer, R.; Nidetzky, B. NMR study of 13C-kinetic isotope effects at 13C natural abundance to characterize oxidations and an enzyme-catalyzed reduction. Tetrahedron Lett. 2006, 47, 4045–4050. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Honegger, P.; Widhalm, M. Formaldehyde-1,1″-(phenylphosphinidenesulfide)bis[(2S)-2-[(1R)-1-(methylamino)-ethyl]]ferrocene-aminal. Molbank 2019, 2019, M1090. https://doi.org/10.3390/M1090
Honegger P, Widhalm M. Formaldehyde-1,1″-(phenylphosphinidenesulfide)bis[(2S)-2-[(1R)-1-(methylamino)-ethyl]]ferrocene-aminal. Molbank. 2019; 2019(4):M1090. https://doi.org/10.3390/M1090
Chicago/Turabian StyleHonegger, Philipp, and Michael Widhalm. 2019. "Formaldehyde-1,1″-(phenylphosphinidenesulfide)bis[(2S)-2-[(1R)-1-(methylamino)-ethyl]]ferrocene-aminal" Molbank 2019, no. 4: M1090. https://doi.org/10.3390/M1090
APA StyleHonegger, P., & Widhalm, M. (2019). Formaldehyde-1,1″-(phenylphosphinidenesulfide)bis[(2S)-2-[(1R)-1-(methylamino)-ethyl]]ferrocene-aminal. Molbank, 2019(4), M1090. https://doi.org/10.3390/M1090