(E)-4-(3-Phenylisoxazol-5-yl)but-3-en-2-one


Abstract
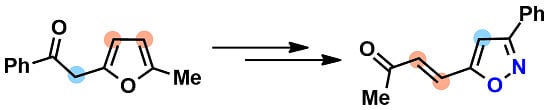
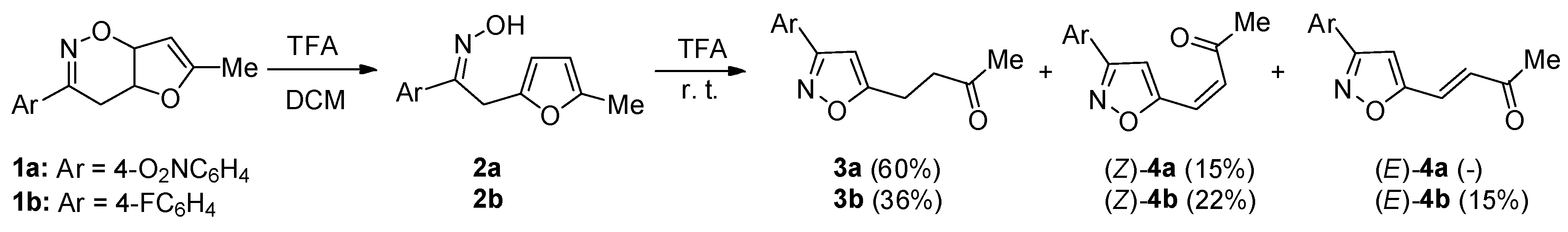
1. Introduction
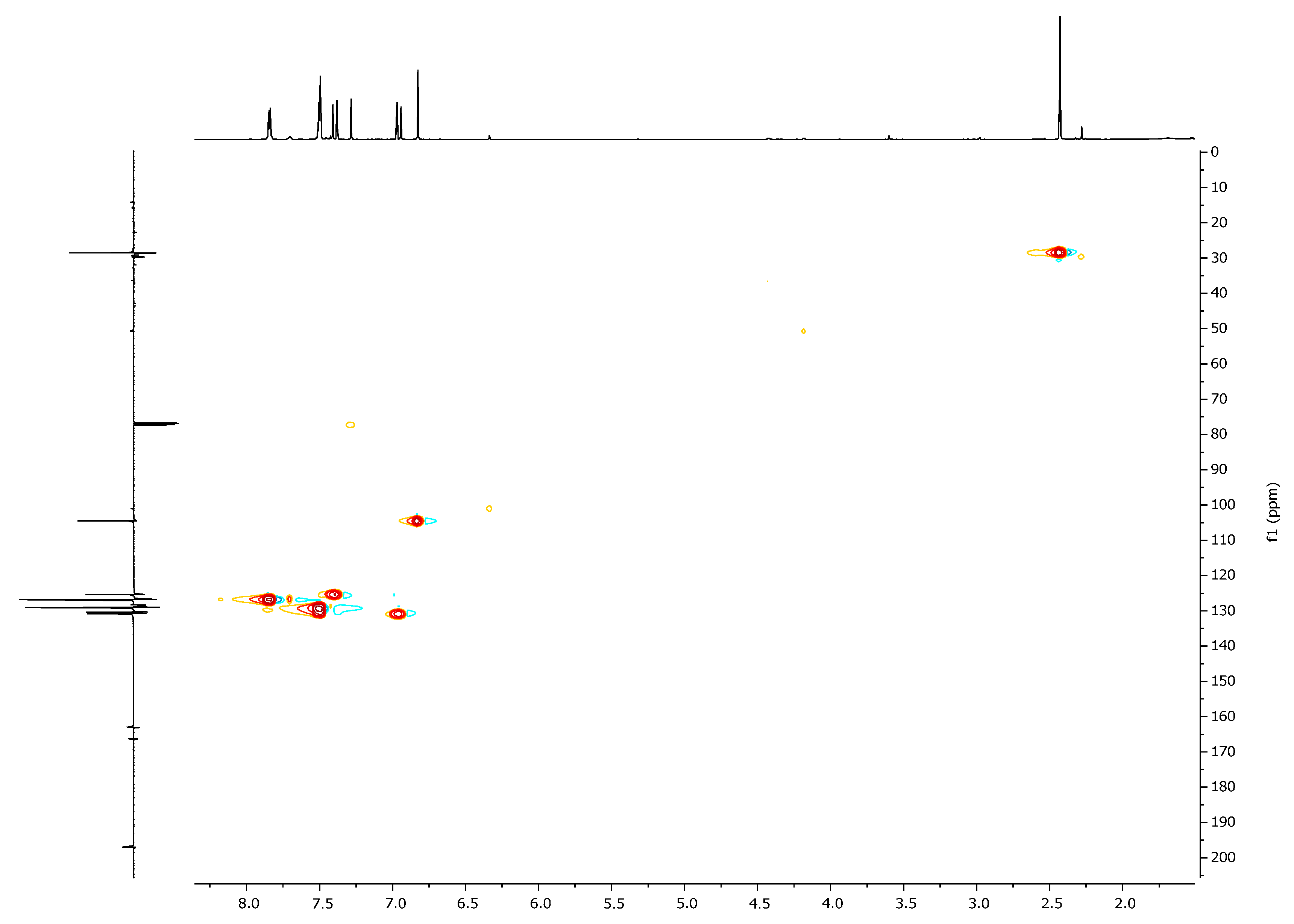
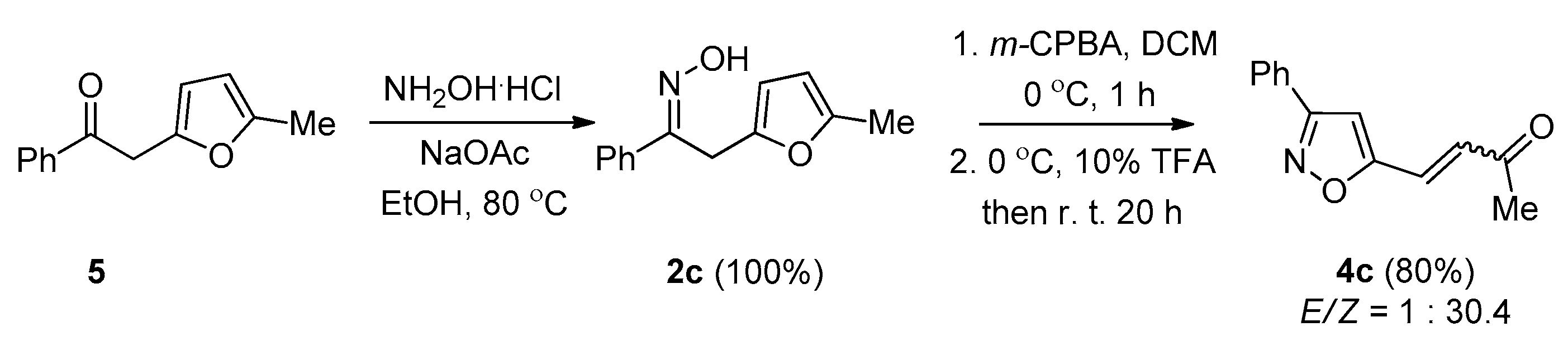
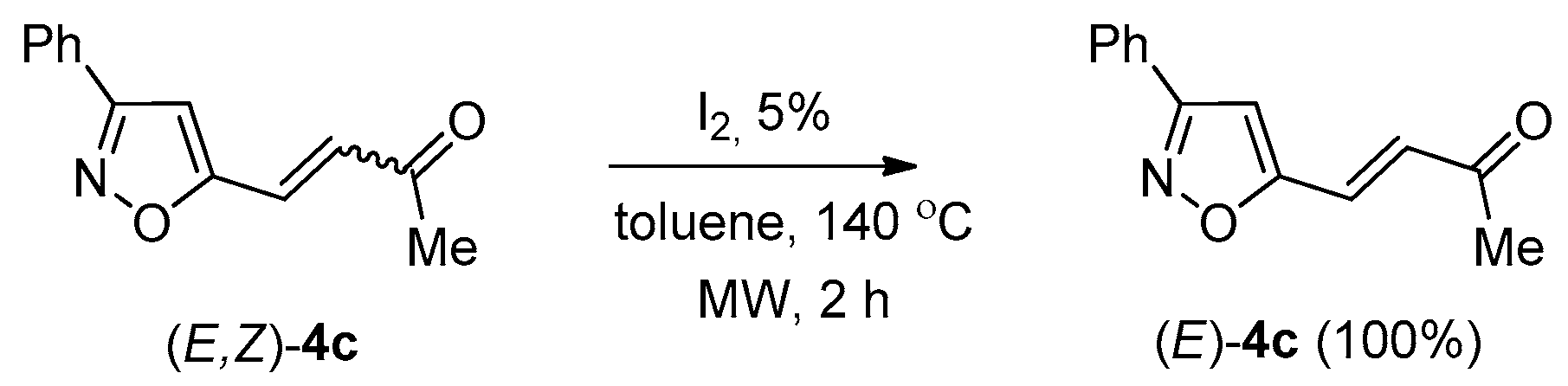
2. Results and Discussion
3. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sysak, A.; Obmińska-Mrukowicz, B. Isoxazole ring as a useful scaffold in a search for new therapeutic agents. Eur. J. Med. Chem. 2017, 137, 292–309. [Google Scholar] [CrossRef] [PubMed]
- Rajanarendar, E.; Nagi Reddy, M.; Rama Krishna, S.; Govardhan Reddy, K.; Reddy, Y.N.; Rajam, M.V. Design, synthesis, in vitro antimicrobial and anticancer activity of novel methylenebis-isoxazolo [4,5-b]azepines derivatives. Eur. J. Med. Chem. 2012, 50, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Güiza, F.M.; Duarte, Y.B.; Mendez-Sanchez, S.C.; Bohórquez, A.R.R. Synthesis and in vitro evaluation of substituted tetrahydroquinoline-isoxazole hybrids as anticancer agents. Med. Chem. Res. 2019, 28, 1182–1196. [Google Scholar] [CrossRef]
- Wang, H.; Ma, Y.; Lin, Y.; Liu, J.; Chen, R.; Xu, B.; Liang, Y. An Isoxazole derivative SHU00238 suppresses colorectal cancer growth through miRNAs regulation. Molecules 2019, 24, 2335. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Zhang, Y.; Wang, C.; Zhang, M.; Xiang, Q.; Wang, J.; Wang, A.; Li, C.; Zhang, C.; Zou, L.; et al. Benzoxazinone-containing 3,5-dimethylisoxazole derivatives as BET bromodomain inhibitors for treatment of castration-resistant prostate cancer. Eur. J. Med. Chem. 2018, 152, 542–559. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.M.M.; Faria, N.; Iley, J.; Coles, S.J.; Hursthouse, M.B.; Martins, M.L.; Moreira, R. Reaction of naphthoquinones with substituted nitromethanes. Facile synthesis and antifungal activity of naphtho[2,3-d]isoxazole-4,9-diones. Bioorg. Med. Chem. Lett. 2010, 20, 193–195. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.F.; Tückmantel, W.; Eaton, J.B.; Caldarone, B.; Fedolak, A.; Hanania, T.; Brunner, D.; Lukas, R.J.; Kozikowski, A.P. Identification of novel α4β2-nicotinic acetylcholine receptor (nAChR) agonists based on an isoxazole ether scaffold that demonstrate antidepressant-like activity. J. Med. Chem. 2012, 55, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Padmaja, A.; Rajasekhar, C.; Muralikrishna, A.; Padmavathi, V. Synthesis and antioxidant activity of oxazolyl/thiazolylsulfonylmethyl pyrazoles and isoxazoles. Eur. J. Med. Chem. 2011, 46, 5034–5038. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Chang, M.; Villegas-Estrada, A.; Franzblau, S.G.; Möllmann, U.; Miller, M.J. Structure–activity relationship of new anti-tuberculosis agents derived from oxazoline and oxazole benzyl esters. Eur. J. Med. Chem. 2010, 45, 1703–1716. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, Y.; Miao, W.; Qu, J. Synthesis, structures, and herbicidal activity of isoxazole derivatives. J. Heterocycl. Chem. 2010, 47, 1310–1316. [Google Scholar] [CrossRef]
- Upadhyay, A.; Gopal, M.; Srivastava, C.; Pandey, N.D. Isoxazole derivatives as a potential insecticide for managing Callosobruchus chinensis. J. Pestic. Sci. 2010, 35, 464–469. [Google Scholar] [CrossRef]
- Alves, A.J.S.; Lopes, S.M.M.; Henriques, M.S.C.; Paixão, J.A.; Pinho e Melo, T.M.V.D. Hetero-Diels–Alder and ring-opening reactions of furans applied to the synthesis of functionalized heterocycles. Eur. J. Org. Chem. 2017, 27, 4011–4025. [Google Scholar] [CrossRef]
- Voskienė, A.; Mickevičius, V. Cyclization of chalcones to isoxazole and pyrazole derivatives. Chem. Heterocycl. Compd. 2009, 45, 1485–1488. [Google Scholar]
- For application of some related systems through the functionalization of enone fragment, see: Trushkov, I.V.; Uchuskin, M.U.; Abaev, V.T.; Serdyuk, O.V. Indolylvinyl ketones: Building blocks for the synthesis of natural products and bioactive compounds. Synthesis 2019, 51, 787–815.
- Merino, P.; Tejero, T.; Delso, J.I.; Matute, R. Furan oxidations in organic synthesis: Recent advances and applications. Curr. Org. Chem. 2007, 11, 1076–1091. [Google Scholar] [CrossRef]
- Makarov, A.S.; Uchuskin, I.V.; Trushkov, I.V. Furan oxidation reactions in the total synthesis of natural products. Synthesis 2018, 50, 3059–3086. [Google Scholar]
- Dugave, C.; Demange, L. Cis−Trans isomerization of organic molecules and biomolecules: implications and applications. Chem. Rev. 2003, 103, 2475–2532. [Google Scholar] [CrossRef] [PubMed]
- Sawengngen, N.; Chalikidi, P.N.; Araby, S.; Hampel, F.; Gmeiner, P.; Serdyuk, O.V. Synthesis of pyrazolylvinyl ketones from furan derivatives. Org. Biomol. Chem. 2019, 17, 4850–4855. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

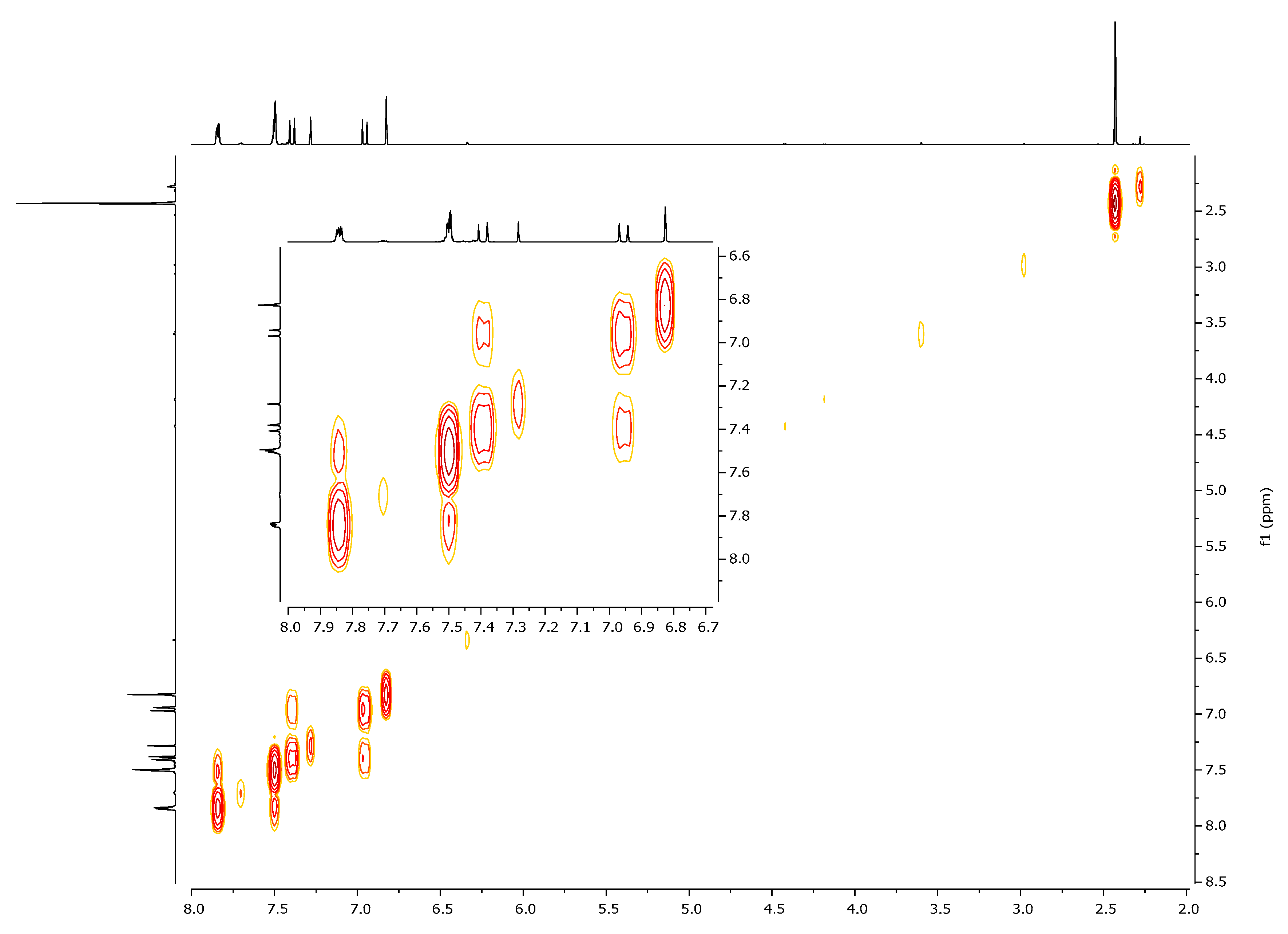
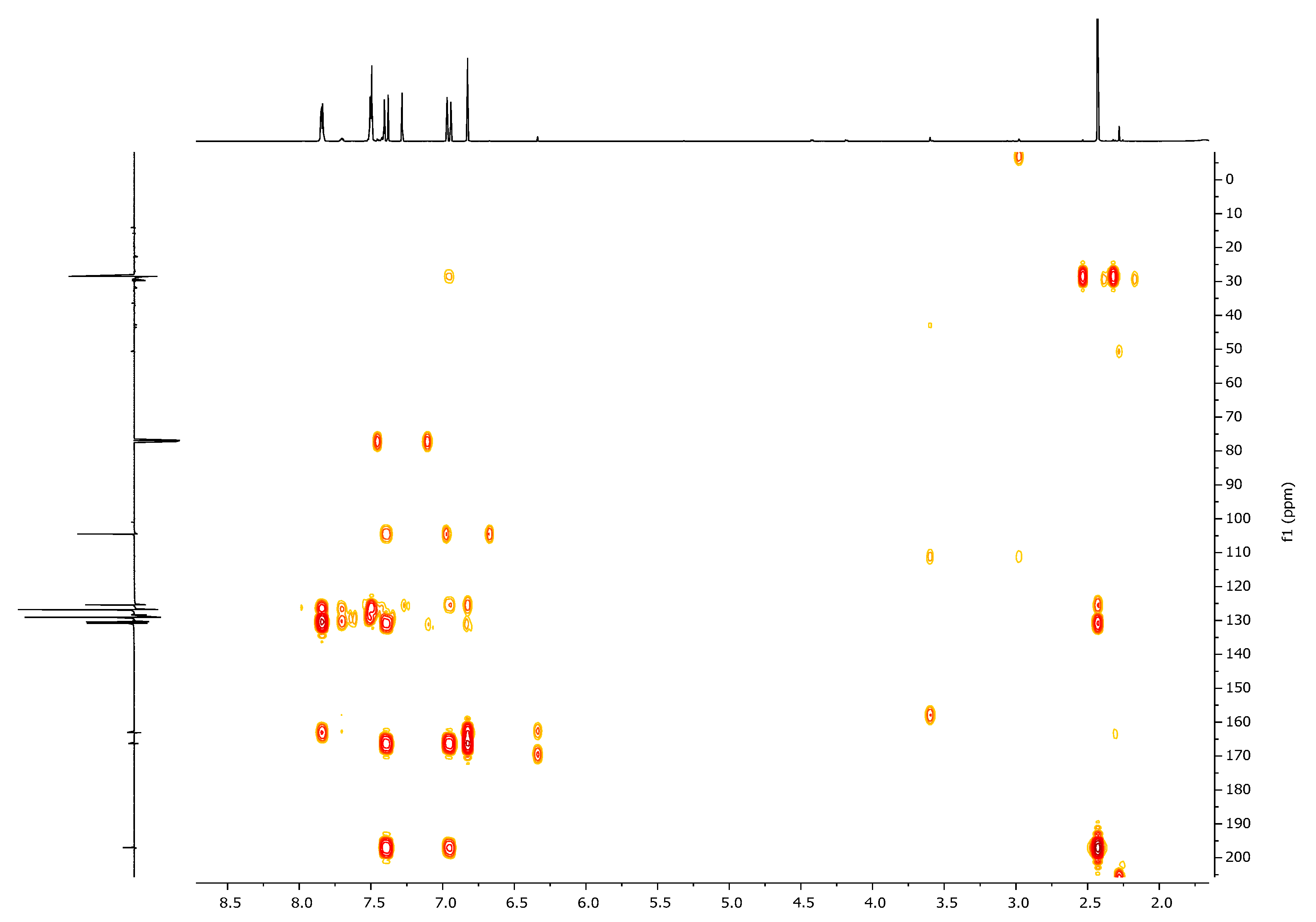
| 1H, δ, ppm | 13C, δ, ppm | |
|---|---|---|
| HMQC | HMBC | |
| 7.40 (β-H) | 125.4 | 104.5, 130.8, 166.3, 197.0 (C=O) |
| 6.95 (α-H) | 130.8 | 28.5, 125.4, 166.3, 197.0 (C=O) |
| 6.80 (HIso) | 104.5 | 125.4, 163.1, 166.3 |
| 2.41 (CH3) | 28.5 | 130.8, 197.0 (C=O) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sawengngen, N.; Kolodina, A.A.; Serdyuk, O.V. (E)-4-(3-Phenylisoxazol-5-yl)but-3-en-2-one. Molbank 2019, 2019, M1081. https://doi.org/10.3390/M1081
Sawengngen N, Kolodina AA, Serdyuk OV. (E)-4-(3-Phenylisoxazol-5-yl)but-3-en-2-one. Molbank. 2019; 2019(3):M1081. https://doi.org/10.3390/M1081
Chicago/Turabian StyleSawengngen, Nattawut, Alexandra A. Kolodina, and Olga V. Serdyuk. 2019. "(E)-4-(3-Phenylisoxazol-5-yl)but-3-en-2-one" Molbank 2019, no. 3: M1081. https://doi.org/10.3390/M1081
APA StyleSawengngen, N., Kolodina, A. A., & Serdyuk, O. V. (2019). (E)-4-(3-Phenylisoxazol-5-yl)but-3-en-2-one. Molbank, 2019(3), M1081. https://doi.org/10.3390/M1081