2-{[Bis(propan-2-yl)carbamothioyl]sulfanyl}acetic acid

 ,
,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
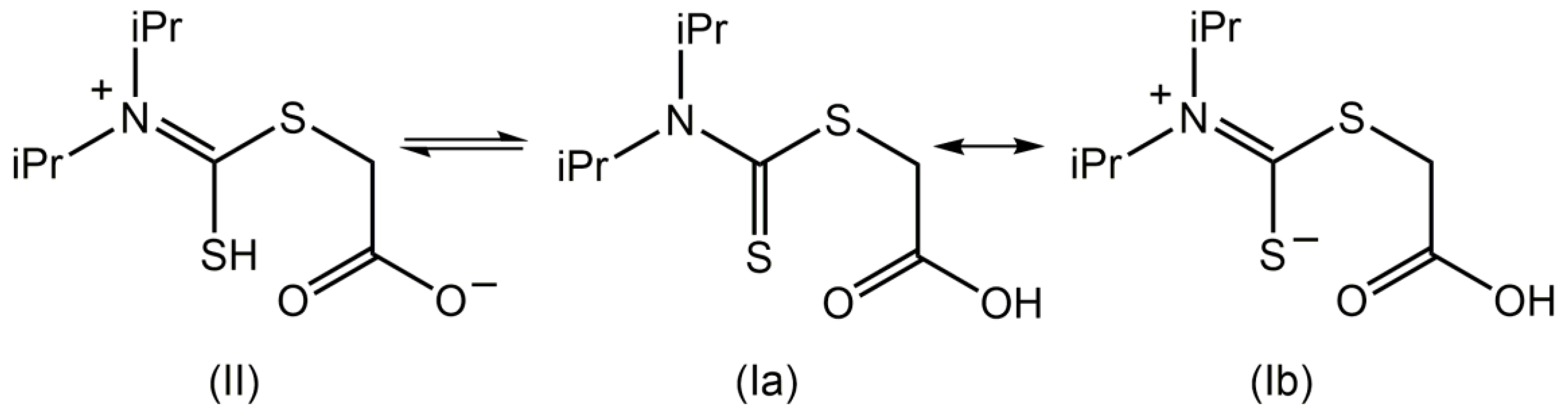
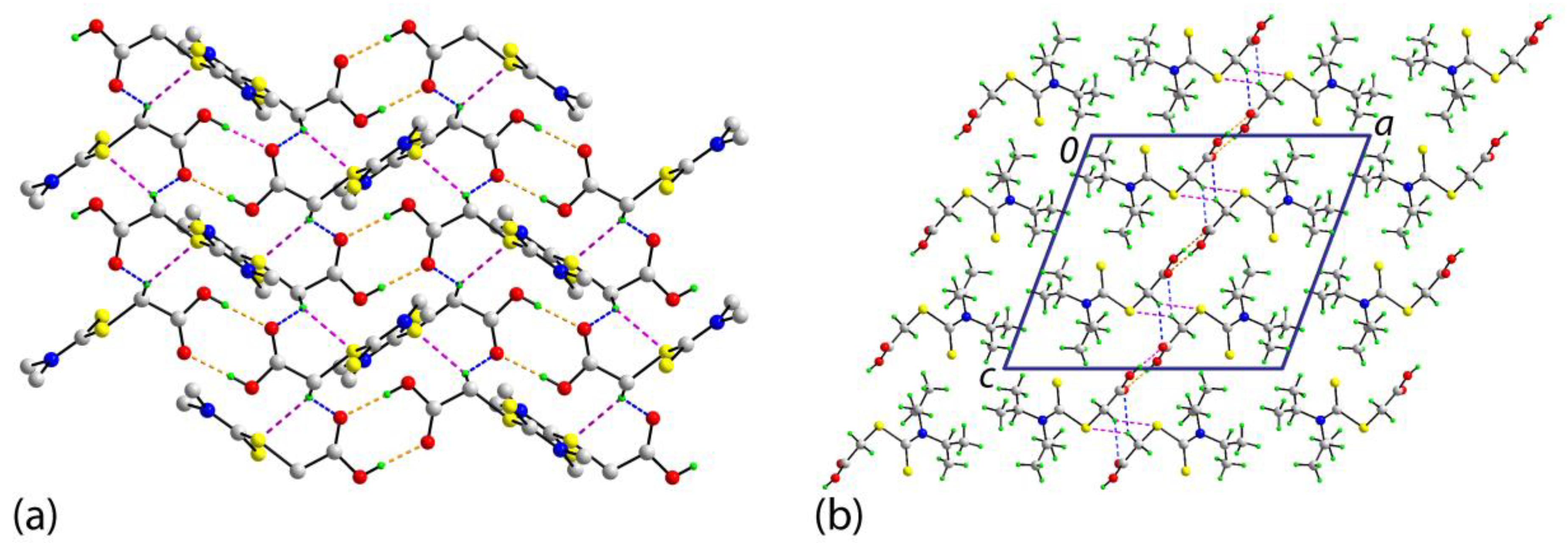
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthesis and Characterization
3.3. Crystallography
3.4. Computational Studies
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Anasamy, T.; Thy, C.K.; Lo, K.M.; Chee, C.F.; Yeap, S.K.; Kamalidehghan, B.; Chung, L.Y. Tribenzyltin carboxylates as anticancer drug candidates: Effect on the cytotoxicity, motility and invasivenessof breast cancer cell lines. Eur. J. Med. Chem. 2017, 125, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Lo, K.M.; Tiekink, E.R.T. Crystal structure of bis[(μ3-oxido)-(μ2-(N,N-diisopropylthiocarbamoylthio)acetato-κ2O,O’)-((N,N-diisopropylthiocarbamoylthio)acetato-κO)-bis(di-4-methylbenzyl-tin(IV))], C100H136N4O10S8Sn4. Z. Kristallogr. New Cryst. Struct. 2019, 234. in press. [Google Scholar] [CrossRef]
- Gielen, M.; Tiekink, E.R.T. 50Sn Tin compounds and their therapeutic potential. In Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The Use of Metals in Medicine; Gielen, M., Tiekink, E.R.T., Eds.; John Wiley & Sons Ltd.: Chichester, UK, 2005; Chapter 22; pp. 421–439. [Google Scholar]
- Tiekink, E.R.T. Tin dithiocarbamates: Applications and structures. Appl. Organomet. Chem. 2008, 22, 533–550. [Google Scholar] [CrossRef]
- Carter, G.A.; Garraway, J.L.; Spencer, D.M.; Wain, R.L. Fungicides. VI. The antifungal activity of certain dithiocarbamic and hydroxydithioformic acid derivatives. Ann. Appl. Biol. 1963, 51, 135–151. [Google Scholar] [CrossRef]
- Nardi, D.; Massarani, E.; Tajana, A.; Degen, L.; Magistretti, M.J. Antibacterial nitrofuran derivatives. I. 5-Nitro-2-furaldehyde semicarbazones and thiosemicarbazones. J. Med. Chem. 1967, 10, 530–533. [Google Scholar] [CrossRef]
- Jensen, K.A.; Anthoni, U.; Kagi, B.; Larsen, C.; Pedersen, C.T. Studies of thioacids and their derivatives. IX. Thiosemicarbazides. Acta Chem. Scand. 1968, 22, 1–50. [Google Scholar] [CrossRef]
- Wakamori, S.; Yoshida, Y.; Ishii, Y. Syntheses and herbicidal activities of dithiocarbamates. I. Benzyl esters of N-substituted dithiocarbamic acids and related compounds. Agric. Biol. Chem. 1969, 33, 1367–1376. [Google Scholar] [CrossRef]
- Bandyopadhyay, P.; Bhattacharya, B.; Majhi, K.; Majee, P.; Sarkar, U.; Seikh, M.M. Benzthiazoline-2-thione (BTT) revisited: An experimental and theoretical endeavor to understand UV-spectra. Chem. Phys. Lett. 2017, 686, 88–96. [Google Scholar] [CrossRef]
- Ng, S.W.; Das, V.G.K. Organotin esters of dithiocarbamylacetic acids. J. Organomet. Chem. 1991, 409, 143–156. [Google Scholar] [CrossRef]
- Le-Thanh, H.; Vocelle, D. 1H NMR studies of proton transfer in Schiff base and carboxylic acid systems. Can. J. Chem. 1990, 68, 1909–1916. [Google Scholar] [CrossRef]
- Couperus, P.A.; Clague, A.D.H.; van Dongen, J.P.C.M. Carbon-13 chemical shifts of some model carboxylic acids and esters. Org. Mag. Res. 1978, 11, 1590–1597. [Google Scholar] [CrossRef]
- Takeda, Y.; Tanaka, T. Three rotational isomers of se-methyl N,N-di-isopropyldiselenocarbamate and the sulphur analogue found in low temperature 1H NMR spectra. Org. Mag. Reson. 1975, 7, 107–108. [Google Scholar] [CrossRef]
- Liljefors, T.; Sandström, J. The gear effect. VII†—Conformational analysis of methyl N,N-diisopropylcarbamate, its thiol and diseleno analogues. An experimental proof for a two-step conformational interconversion mechanism. Org. Mag. Reson. 1977, 9, 276–280. [Google Scholar] [CrossRef]
- Vogel, L.; Wonner, P.; Huber, S.M. Chalogen bonding: An overview. Angew. Chem. Int. Ed. 2019, 58, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.J.; Mckinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. Crystal Explorer v17.; The University of Western Australia: Crawley, Australia, 2017. [Google Scholar]
- Tan, S.L.; Jotani, M.M.; Tiekink, E.R.T. Utilizing Hirshfeld surface calculations, non-covalent interaction (NCI) plots and the calculation of interaction energies in the analysis of molecular packing. Acta Crystallogr. E 2019, 75, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.M.; Ng, S.W. 2-(4-Morpholine-carbothio-ylsulfan-yl)-acetic acid. Acta Crystallogr. E 2010, 66, o1078. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlis PRO; Rigaku Corporation: Oxford, UK, 2017. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. DIAMOND.; Crystal Impact GbR: Bonn, Germany, 2006. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 16; Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView; Version 6; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian-basis sets for molecular calculation. 1. 2nd row atoms, Z=11-18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.M.; Azizan, A.H.S.; Lo, K.M.; Tan, S.L.; Tiekink, E.R.T. 2-{[Bis(propan-2-yl)carbamothioyl]sulfanyl}acetic acid. Molbank 2019, 2019, M1082. https://doi.org/10.3390/M1082
Lee SM, Azizan AHS, Lo KM, Tan SL, Tiekink ERT. 2-{[Bis(propan-2-yl)carbamothioyl]sulfanyl}acetic acid. Molbank. 2019; 2019(4):M1082. https://doi.org/10.3390/M1082
Chicago/Turabian StyleLee, See Mun, Ainnul Hamidah Syahadah Azizan, Kong Mun Lo, Sang Loon Tan, and Edward R. T. Tiekink. 2019. "2-{[Bis(propan-2-yl)carbamothioyl]sulfanyl}acetic acid" Molbank 2019, no. 4: M1082. https://doi.org/10.3390/M1082
APA StyleLee, S. M., Azizan, A. H. S., Lo, K. M., Tan, S. L., & Tiekink, E. R. T. (2019). 2-{[Bis(propan-2-yl)carbamothioyl]sulfanyl}acetic acid. Molbank, 2019(4), M1082. https://doi.org/10.3390/M1082