2-[2-Methyl-5-phenyl-1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-3-yl]-2-oxo-N-(pyridin-4-yl) acetamide


Abstract
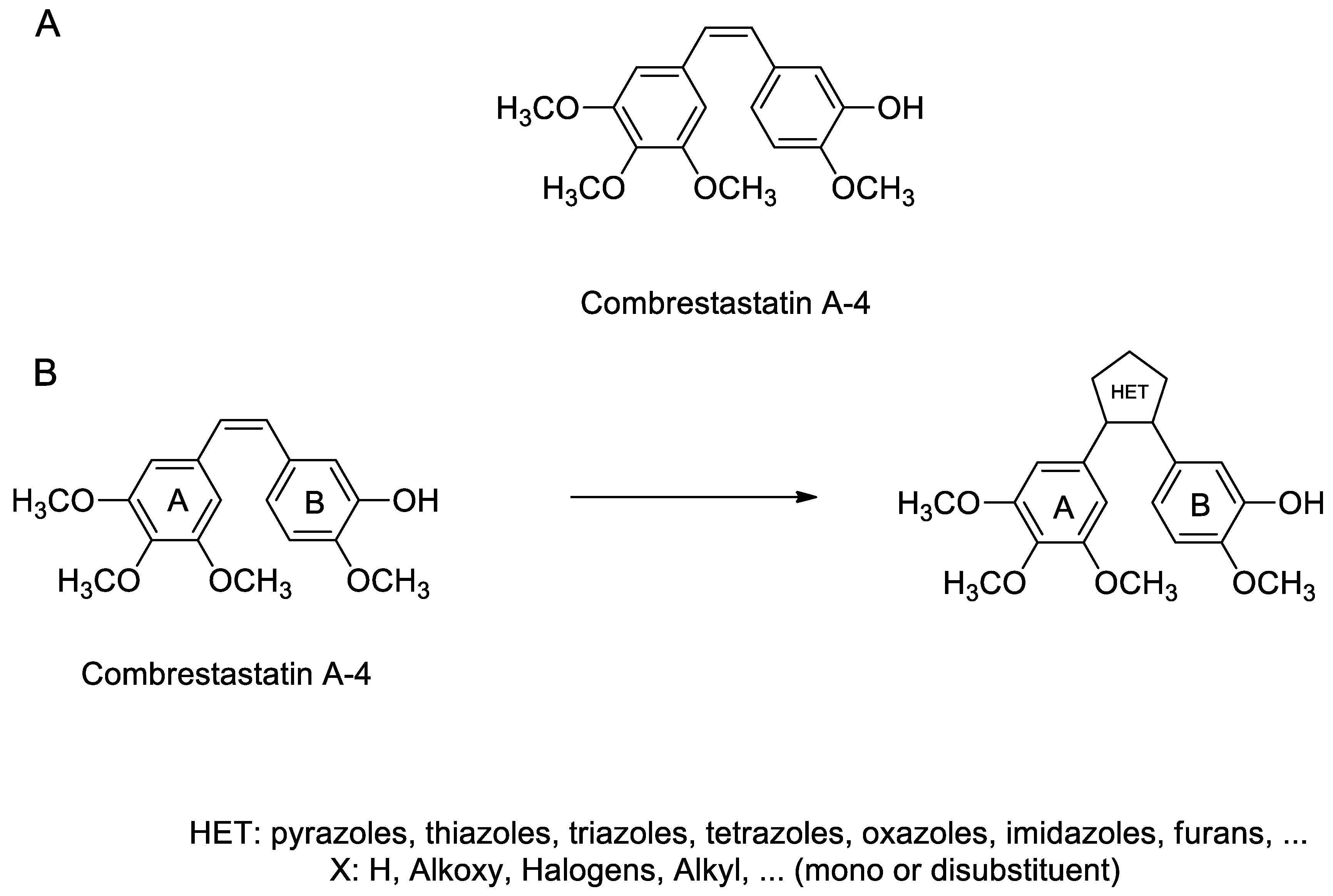
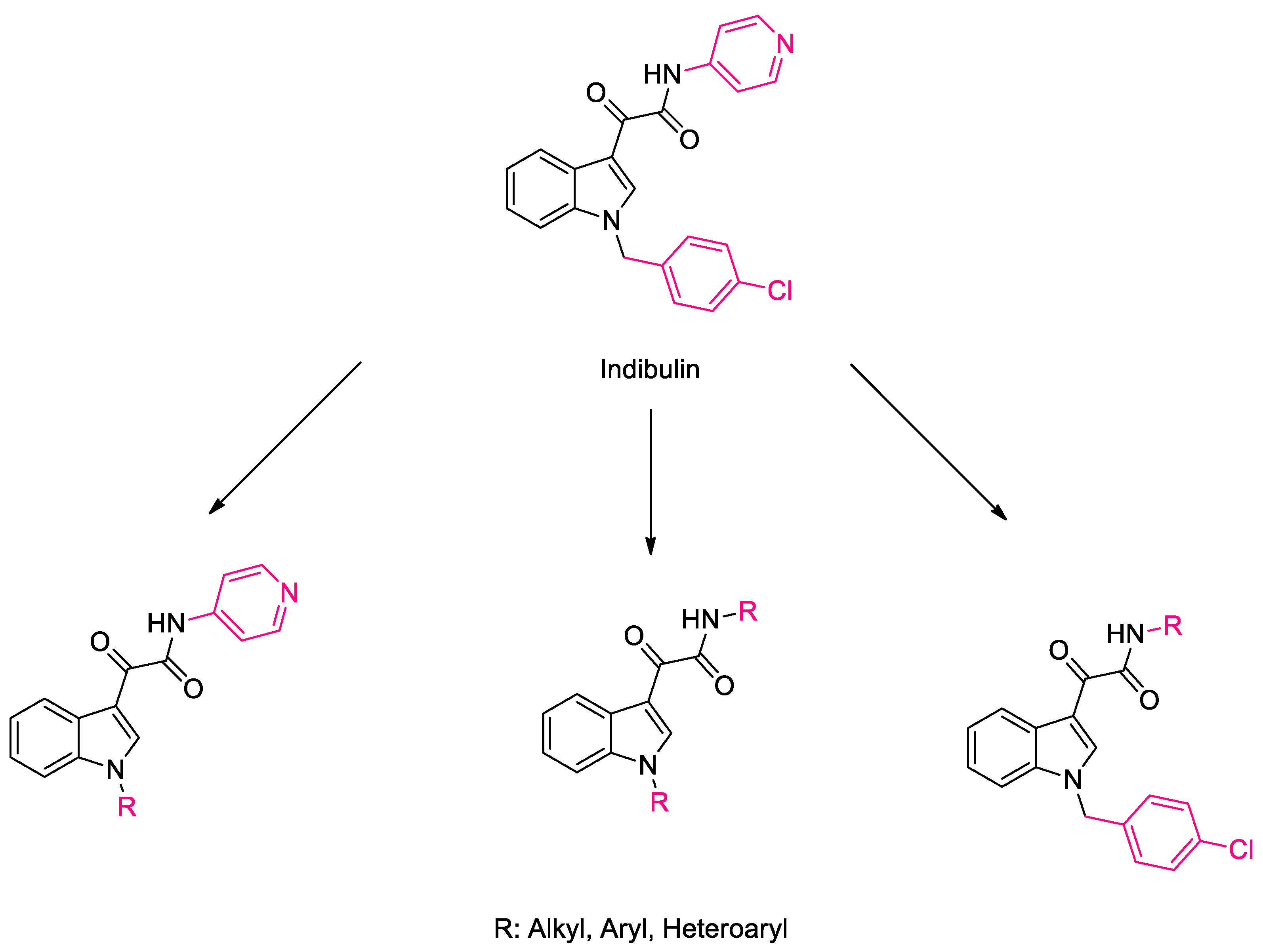
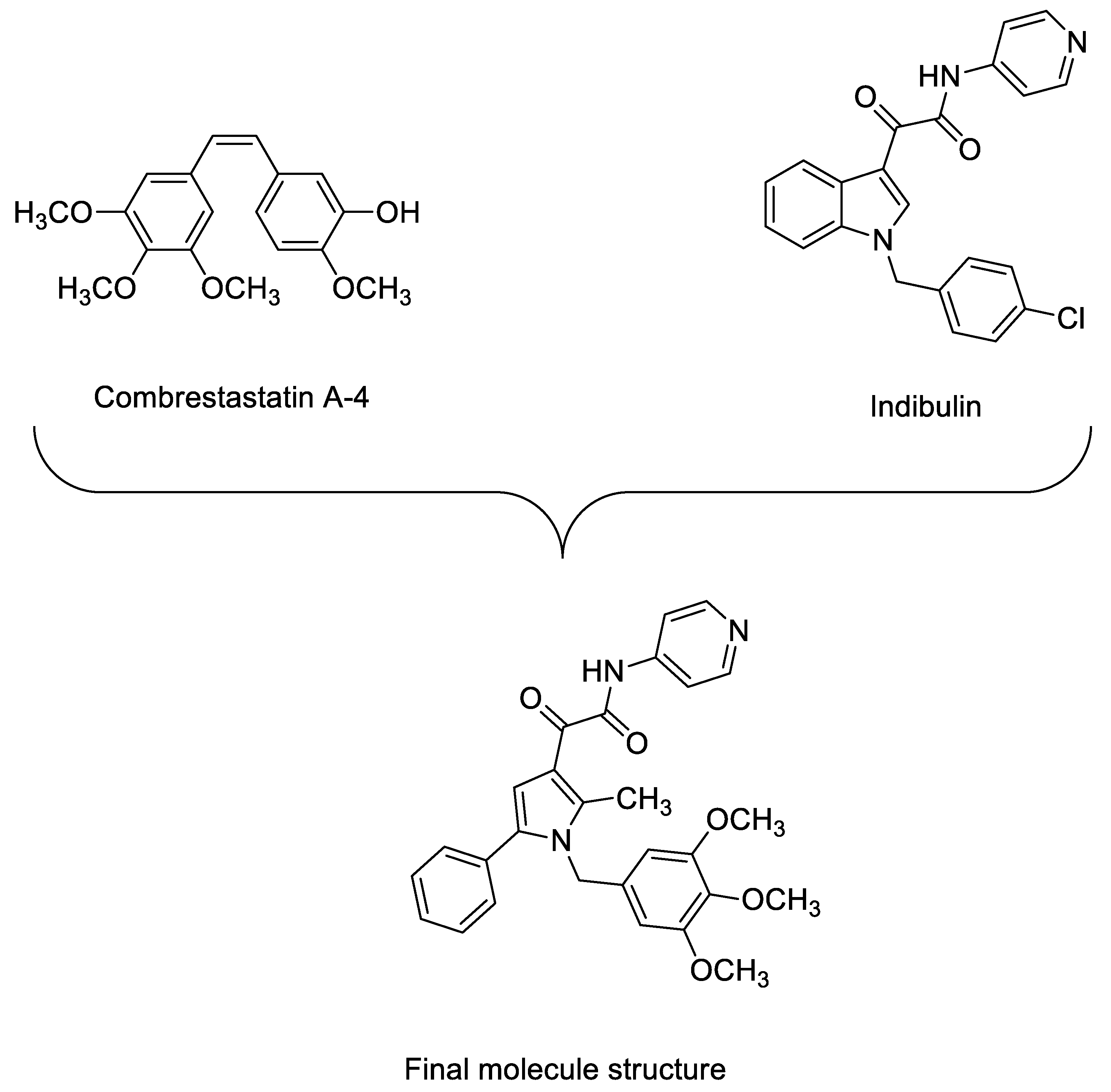
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Anticancer Activity
3. Materials and Methods
3.1. General
3.2. Chemistry
3.2.1. Procedure for the Preparation of 2-Methyl-5-phenyl-1-(3,4,5-trimethoxyphenyl)-1H-pyrrole (3)
3.2.2. Procedure for the Preparation of 2-[2-Methyl-5-phenyl-1-(3,4,5-trimethoxyphenyl) 1H-pyrrol-3-yl]-2-oxo-N-(pyridin-4-yl) acetamide (4)
3.3. Biological Evaluation
3.3.1. Cell Culture
3.3.2. Cytotoxicity Evaluation by MTT Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Prakasham, A.P.; Saxena, A.K.; Luqman, S.; Chanda, D.; Kaur, T.; Gupta, A.; Yadav, D.K.; Chanotiya, C.S.; Shanker, K.; Khan, F.; et al. Synthesis and anticancer activity of 2-benzylidene indanones through inhibiting tubulin polymerization. Bioorgan. Med. Chem. 2012, 20, 3049–3057. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Mulamoottil, V.A.; Nayak, A.; Ryu, S.; Hou, X.; Song, J.; Yu, J.; Sahu, P.K.; Zhao, L.X.; Choi, S.; et al. Design, Synthesis, and Anticancer Activity of C8-Substituted-4′-Thionucleosides as Potential HSP 90 Inhibitors. Bioorgan. Med. Chem. 2016, 24, 3418–3428. [Google Scholar] [CrossRef] [PubMed]
- Vilanova, C.; Díaz-Oltra, S.; Murga, J.; Falomir, E.; Carda, M.; Redondo-Horcajo, M.; Díaz, J.F.; Barasoain, I.; Marco, J.A. Design and Synthesis of Pironetin Analogue/Colchicine Hybrids and Study of Their Cytotoxic Activity and Mechanisms of Interaction with Tubulin. J. Med. Chem. 2014, 57, 10391–10403. [Google Scholar] [CrossRef] [PubMed]
- Baytas, S.N.; Inceler, N.; Yilmaz, A.; Olgac, A.; Menevse, S.; Banoglu, E.; Hamel, E.; Bortolozzi, R.; Viola, G. Synthesis, biological evaluation and molecular docking studies of trans-indole-3-acrylamide derivatives, a new class of tubulin polymerization inhibitors. Bioorgan. Med. Chem. 2014, 22, 3096–3104. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lapidus, R.G.; Sadowska, M.; Edelman, M.J.; Hosmane, R.S. Synthesis, anticancer activity, and SAR analyses of compounds containing the 5:7-fused 4,6,8-triaminoimidazo[4,5-e] [1,3]diazepine ring system. Bioorgan. Med. Chem. 2016, 24, 2595–602. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Perreault, M.; Maltais, R.; Roy, J.; Dutour, R.; Poirier, D. Design of a Mestranol 2-N-Piperazino-Substituted Derivative Showing Potent and Selective in vitro and in vivo Activities in MCF-7 Breast Cancer Models. ChemMedChem 2017, 12, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Rahmani-Nezhad, S.; Safavi, M.; Pordeli, M.; Ardestani, S.K.; Khosravani, L.; Pourshojaei, Y.; Mahdavi, M.; Emami, S.; Foroumadi, A.; Shafiee, A. Synthesis, in vitro cytotoxicity and apoptosis inducing study of 2-aryl-3-nitro-2Hchromene derivatives as potent anti-breast cancer agents. Eur. J. Med. Chem. 2014, 86, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Ahn, S.; Wang, J.; Chen, J.; Miller, D.D.; Dalton, J.T.; Li, W. Discovery of 4-Aryl-2-benzoyl-imidazoles as Tubulin Polymerization Inhibitor with Potent Antiproliferative Properties. J. Med. Chem. 2013, 56, 3318–3329. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Peng, Y.; Wang, X.I.; Keenan, S.M.; Arora, S.; Welsh, W.J. Highly Potent Triazole-Based Tubulin Polymerization Inhibitors. J. Med. Chem. 2007, 50, 749–754. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Cruz-Lopez, O.; Lopez Cara, C.; Carrion, M.D.; Brancale, A.; Hamel, E.; Chen, L.; Bortolozzi, R.; Basso, G.; et al. Synthesis and Antitumor Activity of 1,5-Disubstituted 1,2,4-Triazoles as Cis-Restricted Combretastatin Analogues. J. Med. Chem. 2010, 53, 4248–4258. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Salvador, M.K.; Preti, D.; Aghazadeh Tabrizi, M.; Brancale, A.; Fu, X.H.; Li, J.; Zhang, S.Z.; Hamel, E.; et al. Synthesis and Evaluation of 1,5-Disubstituted Tetrazoles as Rigid Analogues of Combretastatin A-4 with Potent Antiproliferative and Antitumor Activity. J. Med. Chem. 2012, 55, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Galli, U.; Travelli, C.; Aprile, S.; Arrigoni, E.; Torretta, S.; Grosa, G.; Massarotti, A.; Sorba, G.; Canonico, P.L.; Genazzani, A.A.; et al. Design, synthesis, and biological evaluation of combretabenzodiazepines: A novel class of anti-tubulin agents. J. Med. Chem. 2015, 58, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- Wienecke, A.; Bacher, G. Indibulin, a novel microtubule inhibitor, discriminates between mature neuronal and nonneuronal tubulin. Cancer Res. 2009, 69, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Colley, H.E.; Muthana, M.; Danson, S.J.; Jackson, L.V.; Brett, M.L.; Harrison, J.; Coole, S.F.; Mason, D.P.; Jennings, L.R.; Wong, M.; et al. An orally bioavailable, indole-3-glyoxylamide based series of tubulin polymerization inhibitors showing tumor growth inhibition in a mouse Xenograft model of head and neck cancer. J. Med. Chem. 2015, 58, 9309–9333. [Google Scholar] [CrossRef] [PubMed]
- Marchand, P.; Antoine, M.; Le Baut, G.; Czech, M.; Baasner, S.; Günther, E. Synthesis and structure–activity relationships of N-aryl(indol-3-yl)glyoxamides as antitumor agents. Bioorgan. Med. Chem. 2009, 17, 6715–6727. [Google Scholar] [CrossRef] [PubMed]
- Li, W.T.; Hwang, D.R.; Chen, C.P.; Shen, C.W.; Huang, C.L.; Chen, T.W.; Lin, C.H.; Chang, Y.L.; Chang, Y.Y.; Lo, Y.K.; et al. Synthesis and Biological Evaluation of N-Heterocyclic Indolyl Glyoxylamides as Orally Active Anticancer Agents. J. Med. Chem. 2003, 46, 1706–1715. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, M.; Ghadbeighi, S.; Amirhamzeh, A.; Tabatabai, S.A.; Ostad, S.N.; Shafiee, A.; Amini, M. Synthesis, Molecular Docking Study, and Cytotoxic Activity of 1,3,5-triaryl Pyrazole Derivatives. Lett. Drug Des. Discov. 2016, 13, 121–128. [Google Scholar] [CrossRef]
- Ghadbegi, S.; Ostad, S.N.; Shafiee, A.; Amini, M. Synthesis and Anticancer Activity of 1,3,5-triaryl-1H-pyrazole. Lett. Drug Des. Discov. 2015, 12, 754–759. [Google Scholar] [CrossRef]
- Miralinaghi, P.; Salimi, M.; Amirhamzeh, A.; Norouzi, M.; Kandelousi, H.M.; Shafiee, A.; Amini, M. Synthesis, molecular docking study, and anticancer activity of triaryl-1,2,4-oxadiazole. Med. Chem. Res. 2013, 22, 4253–4262. [Google Scholar] [CrossRef]
- Salehi, M.; Ostad, S.N.; Riazi, G.H.; Assadieskandar, A.; Shavi, T.C.; Shafiee, A.; Amini, M. Synthesis, cytotoxic evaluation, and molecular docking study of 4,5-diaryl-thiazole-2-thione analogs of combretastatin A-4 as microtubule-binding agents. Med. Chem. Res. 2014, 23, 1465–1473. [Google Scholar] [CrossRef]
- Zareian, B.; Ghadbeighi, S.; Amirhamzeh, A.; Ostad, S.N.; Shafiee, A.; Amini, M. Synthesis, Molecular Docking Study, and Cytotoxic Activity of 3,4-diaryl-5-(4-pyridinyl)-1,2,4-oxadiazole. Med. Chem. 2016, 12, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Elahian, F.; Akbari, M.; Ghasemi, M.; Behtooee, N.; Taheri, M.; Amini, M. Synthesis and Anticancer Activity of 2,4,5-triaryl Imidazole Derivatives. Lett. Drug Des. Discov. 2014, 11, 840–843. [Google Scholar] [CrossRef]
- Peloquin, A.J.; Stone, R.L.; Avila, S.E.; Rudico, E.R.; Horn, C.B.; Gardner, K.A.; Ball, D.W.; Johnson, J.E.B.; Iacono, S.T.; Balaich, G.J. Synthesis of 1,3-Diphenyl-6-alkyl/aryl-Substituted Fulvene Chromophores: Observation of π-π Interactions in a 6-Pyrene-Substituted 1,3-Diphenylfulvene. J. Org. Chem. 2012, 77, 6371–6376. [Google Scholar] [CrossRef] [PubMed]
- Assadieskandar, A.; Amini, M.; Ostad, S.N.; Riazi, G.H.; Shavi, T.C.; Shafiei, B.; Shafiee, A. Design, synthesis, cytotoxic evaluation and tubulin inhibitory activity of 4-aryl-5-(3,4,5-trimethoxyphenyl)-2-alkylthio-1H-imidazole derivatives. Bioorgan. Med. Chem. 2013, 21, 2703–2709. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | NIH-3T3 | MCF-7 | T47-D | MDA-MB231 |
|---|---|---|---|---|
| 4 | >100 | 29.1 ± 1.8 | 39.2 ± 3.2 | 27.75 ± 2.6 |
| paclitaxel | 10.2 ± 1.1 | 0.5 ± 0.3 | 1.0 ± 0.7 | 1.5 ± 0.5 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeedian Moghadam, E.; Amini, M. 2-[2-Methyl-5-phenyl-1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-3-yl]-2-oxo-N-(pyridin-4-yl) acetamide. Molbank 2018, 2018, 1002. https://doi.org/10.3390/M1002
Saeedian Moghadam E, Amini M. 2-[2-Methyl-5-phenyl-1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-3-yl]-2-oxo-N-(pyridin-4-yl) acetamide. Molbank. 2018; 2018(3):1002. https://doi.org/10.3390/M1002
Chicago/Turabian StyleSaeedian Moghadam, Ebrahim, and Mohsen Amini. 2018. "2-[2-Methyl-5-phenyl-1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-3-yl]-2-oxo-N-(pyridin-4-yl) acetamide" Molbank 2018, no. 3: 1002. https://doi.org/10.3390/M1002
APA StyleSaeedian Moghadam, E., & Amini, M. (2018). 2-[2-Methyl-5-phenyl-1-(3,4,5-trimethoxyphenyl)-1H-pyrrol-3-yl]-2-oxo-N-(pyridin-4-yl) acetamide. Molbank, 2018(3), 1002. https://doi.org/10.3390/M1002