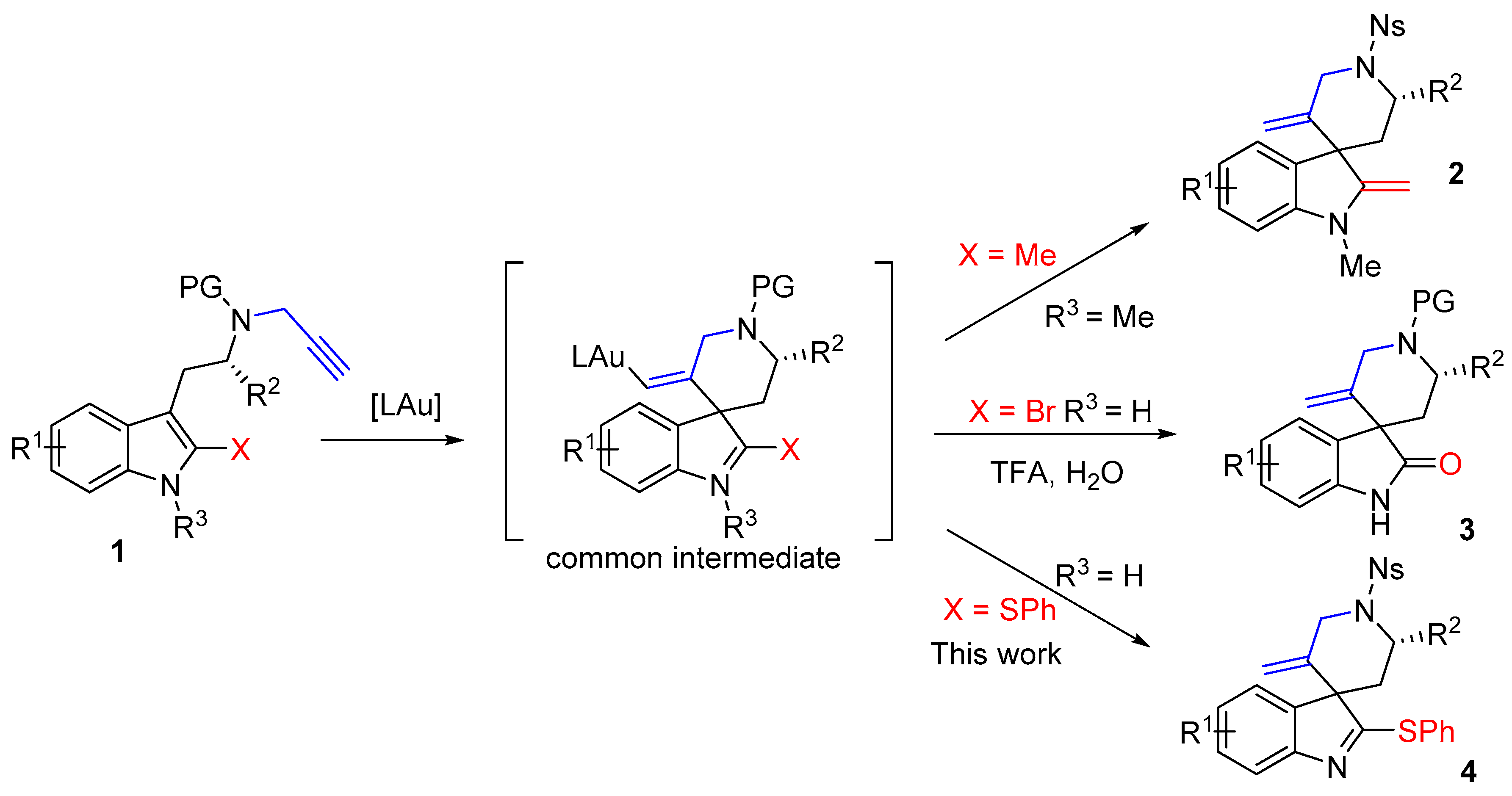
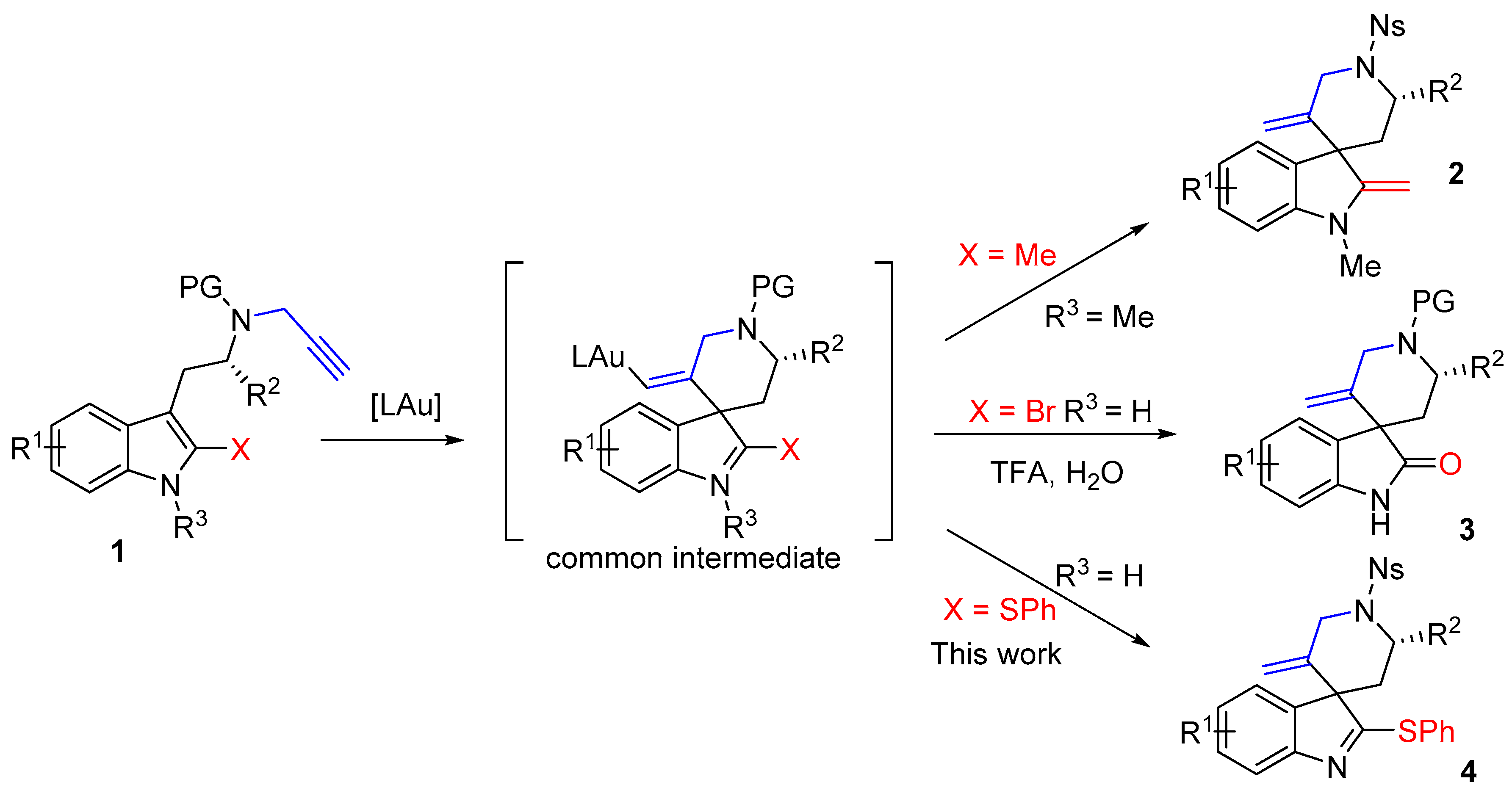
4. Materials and Methods
Reactions were performed using oven dried glasswares under an atmosphere of argon. All separations were carried out under flash-chromatographic conditions on silica gel (Redi Sep prepacked column, 230–400 mesh) at medium pressure (20 psi) with use of a CombiFlash Companion. Reactions were monitored by thin-layer chromatography on Merck silica gel plates (Merck, Darmstadt, Germany) (60 F254 aluminum sheets) which were rendered visible by ultraviolet and spraying with vanillin (15%) + sulfuric acid (2.5%) in EtOH followed by heating. CH2Cl2, DMF and toluene were purchased at the highest commercial quality and used without further purification. Reagent-grade chemicals were obtained from diverse commercial suppliers and used as received. 1H-NMR (500 or 300 MHz) and 13C-NMR (125 or 75 MHz) spectra were recorded on Bruker Avance spectrometers (Bruker, Wissembourg, France) at 298 K unless otherwise stated. Chemical shifts are given in ppm (δ) and are referenced to the internal solvent signal or to TMS used as an internal standard. Coupling constants J are given in Hz. Carbon multiplicities were determined by DEPT135 experiment. Diagnostic correlations were obtained by two-dimensional COSY, HSQC and NOESY experiments. Infrared spectra (IR) were recorded on a Perkin-Elmer FT-IR system (Waltham, MA, USA) using diamond window Dura SamplIR II and the data are reported in reciprocal centimeters (cm−1). Optical rotations were measured on a Anton Paar MCP 300 polarimeter (Anton-Paar, Courtaboeuf, France) at 589 nm. is expressed in deg·mL·g−1·dm−1 and c is expressed in g/100 mL. Melting points were recorded in open capillary tubes on a Buchi B-540 apparatus (Buchi, Rungis, France) and are uncorrected. High resolution mass spectra (HRMS) were recorded using a Micromass LCT Premier XE instrument (Waters, Milford, MA, USA) and were determined by electrospray ionization (ESI).
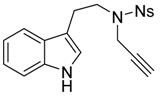
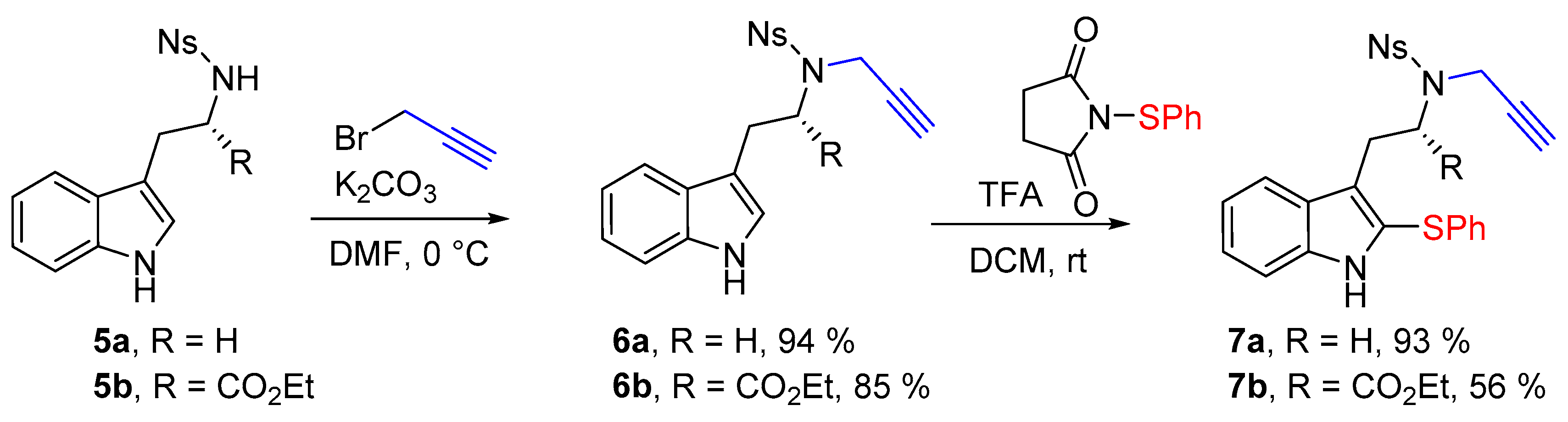
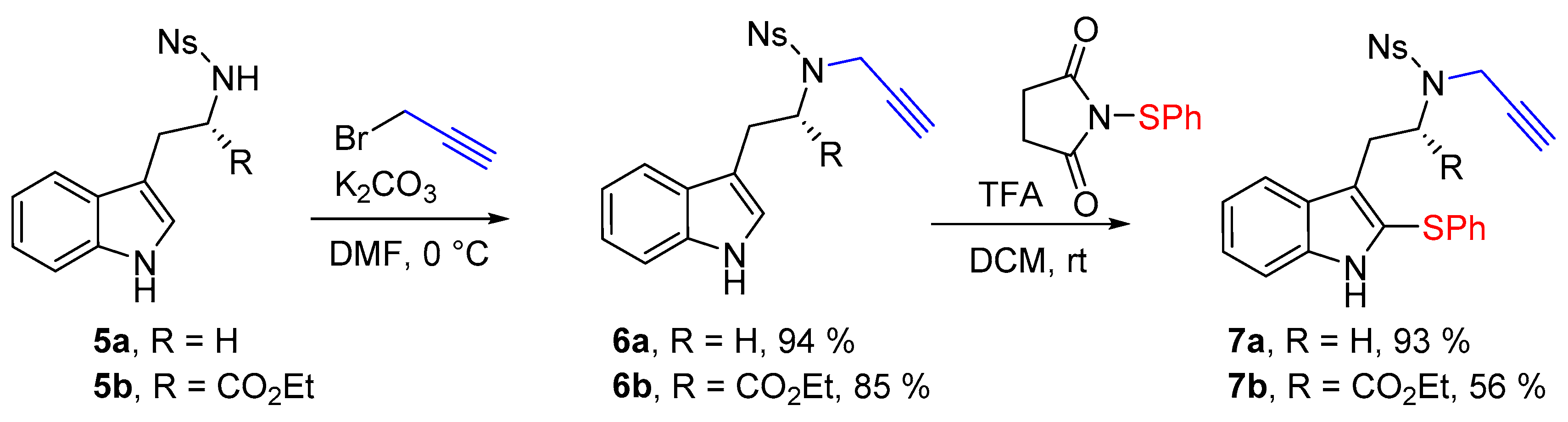
N-[2-(1H-Indol-3-yl)ethyl]-4-nitro-N-(prop-2-yn-1-yl)benzenesulfonamide (6a)
![Molbank 2018 m985 i001]()
N-[2-(1H-Indol-3-yl)ethyl]-4-nitrobenzenesulfonamide (5a) (1.0 g, 2.9 mmol, 1 equiv.) was solubilized in dry dimethylformamide (30 mL) under argon, and K2CO3 (483 mg, 3.5 mmol, 1.2 equiv.) and propargyl bromide solution (0.31 mL, 80% w/w solution, 3.5 mmol, 1.2 equiv.) were added sequentially. The reaction mixture was stirred for 4 h at room temperature. Then a saturated aqueous solution of NH4Cl was added before pouring the reaction media in a separatory funnel containing ethyl acetate. After separation of the phases the aqueous phase was extracted twice with ethyl acetate, then the combined organic phases were washed twice with water and brine. The organic phase was then dried over MgSO4 before removing the solvent under reduced pressure. Finally the crude mixture was purified over silica gel column chromatography (eluent 10 to 50% EtOAc/Heptane), furnishing the pure product 6a as an orange foam (1.04 g, 2.71 mmol, 94%).
Rf: 0.40 (50:50 EtOAc:heptane). IR (neat) νmax: 3416, 3285, 3105, 2924, 1526, 1347, 1310, 1160 cm−1. 1H-NMR (CDCl3, 300 MHz): 8.22 (d, J = 8.9 Hz, 2H), 8.02 (brs, 1H), 7.94 (d, J = 8.9 Hz, 2H), 7.57 (d, 7.9 Hz, 1H), 7.34 (dt, J = 8.2 and 0.9 Hz, 1H), 7.20 (ddd, J = 7.9, 7.2 and 1.1 Hz, 1H), 7.12 (ddd, J = 7.9, 7.2 and 1.1 Hz, 1H), 7.07 (d, J = 2.5 Hz, 1H), 4.23 (d, J = 2.4 Hz, 2H), 3.58 (t, J = 7.8 Hz, 2H), 3.10 (t, J = 6.7 Hz, 2H), 2.10 (t, J = 2.5 Hz, 1H). 13C-NMR (CDCl3, 75 MHz): 150.1 (Cq), 144.9 (Cq), 136.4 (Cq), 128.9 (2CH), 127.2 (CH), 124.0 (2CH), 122.5 (CH), 122.4 (CH), 119.8 (CH), 118.6 (CH), 111.9 (Cq), 111.4 (CH), 76.4 (Cq), 74.5 (CH), 47.2 (CH2), 36.8 (CH2), 24.4 (CH2). HRMS (ESI): calcd. for C19H18N3O4S [M + H]+ 384.1018, found 384.1002.
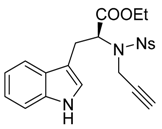
Ethyl-N-[(4-nitrophenyl)sulfonyl]-N-(prop-2-yn-1-yl)-l-tryptophanate (6b)
![Molbank 2018 m985 i002]()
Ethyl [(4-nitrophenyl)sulfonyl]-l-tryptophanate (5b) (415 mg, 1.0 mmol, 1 equiv.) was solubilized in dry dimethylformamide (10 mL) under argon, and K2CO3 (152 mg, 1.2 mmol, 1.2 equiv.) and propargyl bromide solution (0.12 mL, 80% w/w solution, 1.1 mmol, 1.1 equiv.) were added sequentially. The reaction mixture was stirred for 4 h at room temperature. Then a saturated aqueous solution of NH4Cl was added before to pour the reaction media in a separatory funnel containing ethyl acetate. After separation of the phases the aqueous phase was extracted twice with ethyl acetate, then the combined organic phases were washed twice with water and with brine. The organic phase was then dried over MgSO4 before removing the solvent under reduced pressure. Finally the crude mixture was purified over silica gel column chromatography (eluent 10 to 50% EtOAc/Heptane), furnishing the pure product 6b as an orange foam (385 mg, 0.845 mmol, 85%).
IR (neat) νmax: 3370, 3285, 3105, 3070, 1715, 1531, 1353, 1166, 1150, 1093, 887, 856, 748 cm−1. 1H-NMR (CDCl3, 300 MHz): 7.96 (brs, 1H), 7.89 (d, J = 8.8 Hz, 2H), 7.66 (d, J = 8.8 Hz, 2H), 7.50 (d, J = 7.5 Hz, 1H), 7.25–7.22 (m, 1H), 7.17 (td, J = 7.2 and 1.5 Hz, 1H), 7.1 (ddd, J= 7.7, 6.8 and 1.5 Hz, 1H), 6.99 (d, J = 2.5 Hz, 1H), 4.81 (dd, J = 9.6 and 5.3 Hz, 1H), 4.55 (dd, J = 18.8 and 2.5 Hz, 1H), 4.29 (dd, J = 19.8 and 2.6 Hz, 1H), 4.17 (qd, J = 7.2 and 1.7 Hz, 2H), 3.51–3.44 (m, 1H), 3.18 (dd, J = 15.0 and 9.6 Hz, 1H), 2.30 (t, J = 2.4 Hz, 1H), 1.23 (t, J = 7.1 Hz, 3H). 13C-NMR (CDCl3, 75 MHz): 170.4 (Cq), 149.5 (Cq), 145.2 (Cq), 136.2 (Cq), 128.3 (2CH), 126.7 (Cq), 124.0 (CH), 123.2 (2CH), 122.5 (CH), 120.0 (CH), 118.3 (CH), 111.4 (CH), 110.0 (Cq), 79.2 (Cq), 73.4 (CH), 62.0 (CH2), 60.6 (CH), 34.2 (CH2), 26.5 (CH2), 14.2 (CH3). HRMS (ESI): calcd. for C19H18N306S [M − propargyl]− 416.0916, found 416.0908. − 15.5 (c 1.0, CHCl3).
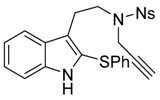
4-Nitro-N-{2-[2-(phenylthio)-1H-indol-3-yl]ethyl}-N-(prop-2-yn-1-yl)benzenesulfonamide (7a)
To a solution of N-[2-(1H-indol-3-yl)ethyl]-4-nitro-N-(prop-2-yn-1-yl)benzenesulfonamide (6a) (383 mg, 1.0 mmol, 1 equiv.) in dry DCM (2 mL) was added 1-(phenylthio)pyrrolidine-2,5-dione (207 mg, 1.0 mmol, 1.0 equiv.), and trifluoroacetic acid (1.15 mL, 15.0 mmol, 15 equiv.). The solution was stirred for 18 h, and the crude media was quenched by a carefull addition of NaHCO3 aqueous saturated solution. The aqueous phase was then extracted two times with DCM. The combined organic phases were dried over MgSO4 and the volatiles were removed under vacuum. The crude mixture was then purified over silica gel column chromatography (eluent 20 to 50% EtOAc/Heptane), furnishing the pure desired product 7a as a yellow amorphous solid (455 mg, 0.93 mmol, 93%).
IR (neat) νmax: 3346, 3272, 3100, 3063, 1603, 1581, 1524, 1444, 1344, 1160, 1090, 855 cm−1. 1H-NMR (CDCl3, 300 MHz): 8.14 (d, J = 9.2 Hz, 2H), 8.08 (brs, 1H), 7.84 (d, J = 9.2 Hz, 2H), 7.61 (d, J = 7.7 Hz, 1H), 7.29–7.27 (m, 1H), 7.25-7.14 (m, 5H), 7.06-7.03 (m, 2H), 4.26 (d, J = 2.5 Hz, 2H), 3.49 (dd, J = 9.0 and 6.3 Hz, 2H), 3.17 (dd, J = 9.1 and 6.8 Hz, 2H), 2.05 (t, J = 2.5 Hz, 1H). 13C-NMR (CDCl3, 75 MHz): 147.2 (Cq), 144.9 (Cq), 137.1 (Cq), 136.8 (Cq), 129.4 (2CH), 128.8 (2CH), 127.5 (Cq), 126.9 (2CH), 126.3 (CH), 124.0 (CH), 124.0 (2CH), 123.0 (Cq), 120.4 (CH), 119.4 (Cq), 119.2 (CH), 111.3 (CH), 76.4 (Cq), 74.5 (CH), 46.9 (CH2), 36.8 (CH2), 24.0 (Cq). HRMS (ESI): calcd. for C25H22N3O4S2 [M + H]+ 492.1046, found 492.1053.
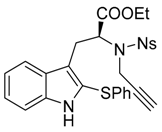
Ethyl-(S)-2-[(4-nitro-N-(prop-2-yn-1-yl)phenyl)sulfonamido)-3-[2-(phenylthio)-1H-indol-3-yl]propanoate (7b)
To solution of ethyl N-[(4-nitrophenyl)sulfonyl]-N-(prop-2-yn-1-yl)-l-tryptophanate (6b) (385 mg, 0.85 mmol, 1 equiv.) in dry DCM (2 mL) was added 1-(phenylthio)pyrrolidine-2,5-dione (184 mg, 0.89 mmol, 1.05 equiv.), and trifluoroacetic acid (0.98 mL, 12.75 mmol, 15 equiv.). The solution was stirred for 18 h, and the crude media was quenched by a careful addition of NaHCO3 aqueous saturated solution. The aqueous phase was then extracted two times with DCM. The combined organic phases were dried over MgSO4 and the volatiles were removed under vacuum. The crude mixture was then purified over silica gel column chromatography (eluent 20 to 50% EtOAc/Heptane), furnishing the pure desired product 7b as a yellow amorphous solid (269 mg, 0.477 mmol, 56%).
IR (neat) νmax: 3372, 3287, 3105, 3070, 2926, 1734, 1717, 1530, 1349, 1164, 1093, 855, 739 cm−1. 1H-NMR (CDCl3, 300 MHz): 8.07 (brs, 1H), 8.00 (d, J = 8.9 Hz, 2H), 7.76 (d, J = 8.9 Hz, 2H), 7.56 (dd, J = 7.9 and 0.9 Hz, 1H), 7.25–7.10 (m, 6H), 7.03–7.00 (m, 2H), 4.91 (t, J = 7.8 Hz, 1H), 4.46 (t, J = 2.2 Hz, 2H), 3.99 (q, J = 7.2 Hz, 2H), 3.57 (dd, J = 14.5 and 7.5 Hz, 1H), 3.31 (dd, J 14.5 and 8.1 Hz, 1H), 2.21 (t, J = 2.5 Hz, 1H), 1.04 (t, J = 7.1 Hz, 3H). 13C-NMR (CDCl3, 75 MHz): 170.1 (Cq), 149.8 (Cq), 145.6 (Cq), 137.0 (Cq), 136.3 (Cq), 129.5 (2CH), 128.7 (2CH), 127.4 (Cq), 127.0 (2CH), 126.4 (CH), 124.1 (CH), 123.6 (2CH + Cq), 120.5 (CH), 119.2 (CH), 117.5 (Cq), 111.3 (CH), 78.7 (Cq), 73.7 (CH), 61.9 (CH2), 59.7 (CH), 34.4 (CH2), 26.1 (CH2), 13.9 (CH3). HRMS (ESI): calcd. for C28H26N3O6S2 [M + H]+ 564.1258, found 464.1268. − 50.4 (c 1.0 CHCl3).
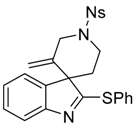
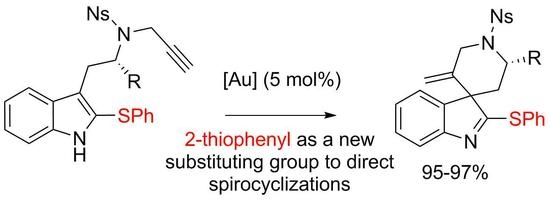
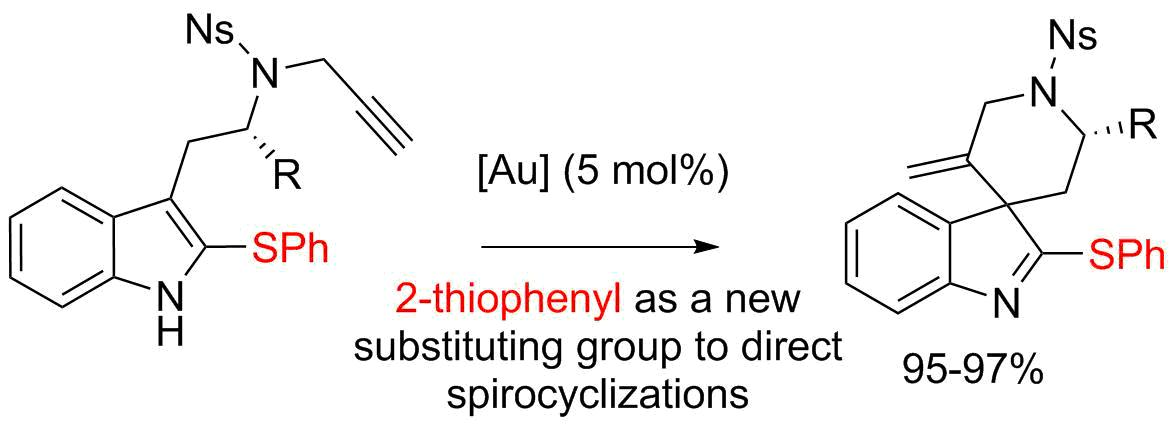
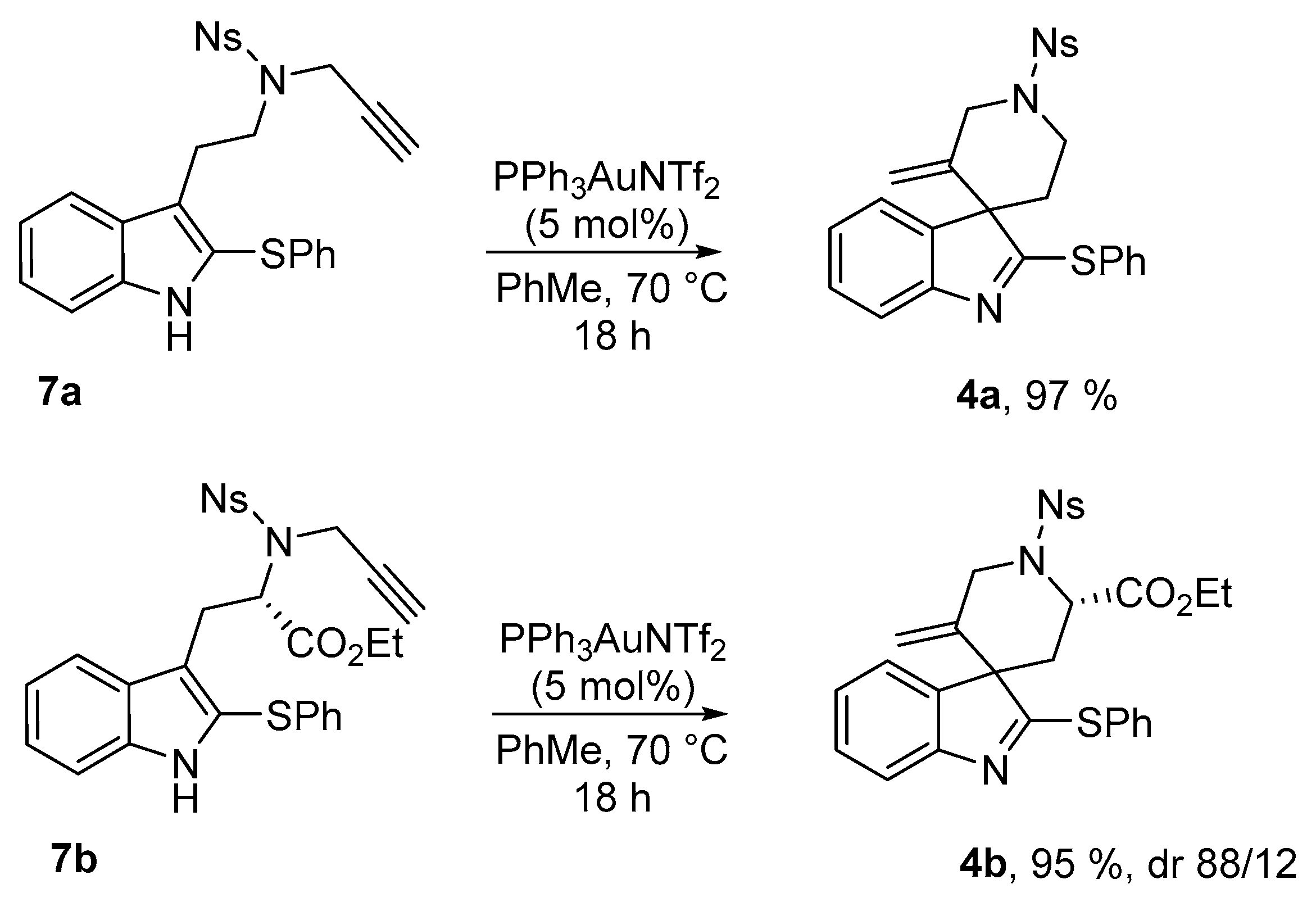
3′-Methylene-1′-[(4-nitrophenyl)sulfonyl]-2-(phenylthio)spiro[indole-3,4′-piperidine] (4a)
4-Nitro-N-{2-[2-(phenylthio)-1H-indol-3-yl]ethyl}-N-(prop-2-yn-1-yl)benzenesulfonamide (7a) (116 mg, 0.236 mmol, 1 equiv.) was pourred in a shlenck tube filled with argon, dry toluene (2.4 mL) and PPh3AuNTf2 (8.7 mg, 0.012 mmol, 5 mol %) were added, and the solution was stirred at 70 °C for 15 h. Then the solvent was evaporated and the crude mixture was purified over silica gel column chromatography (10 to 30% EtOAc:Heptane), furnishing the pure product 4a as a yellowish solid (113 mg, 0.229 mmol, 97%).
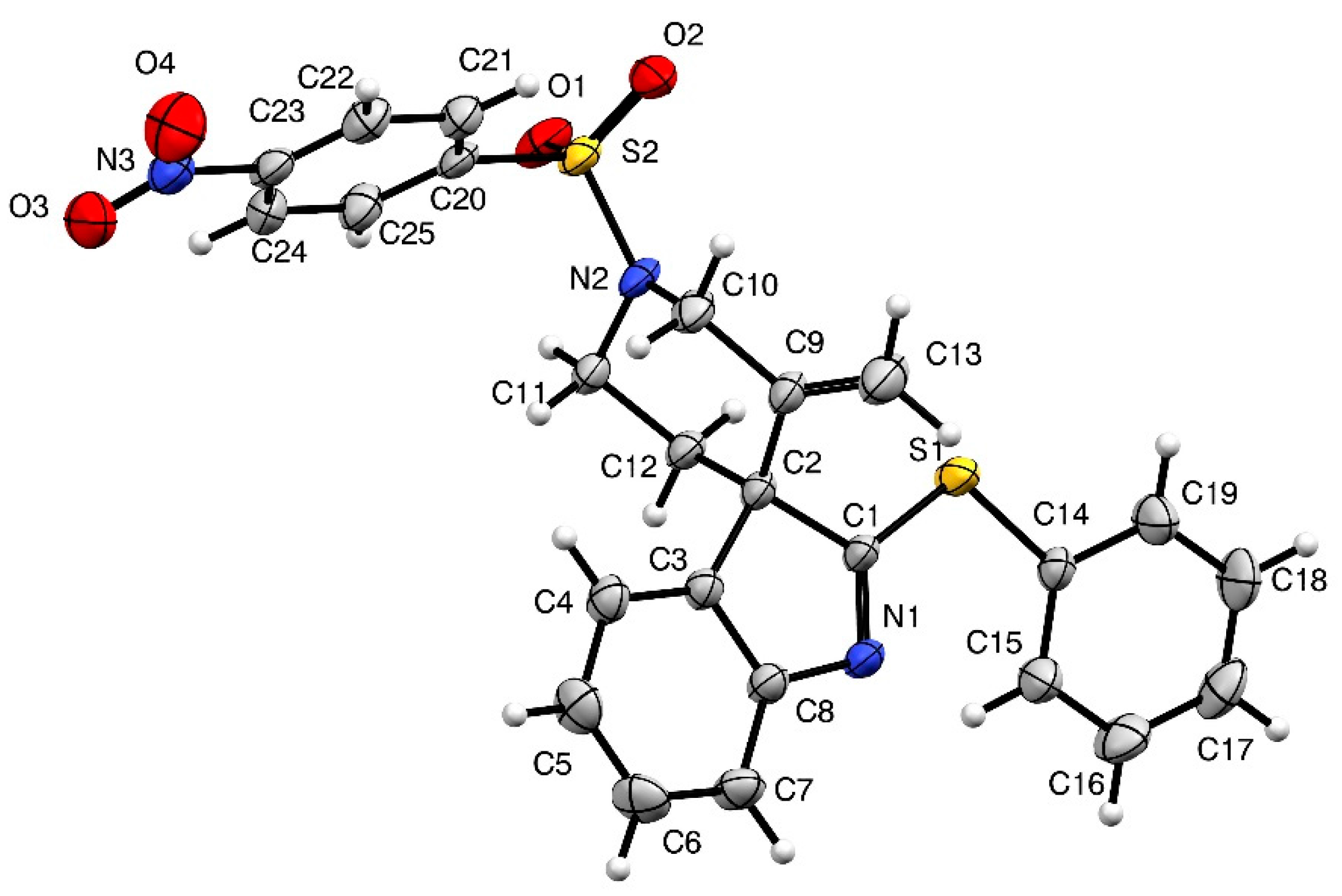
IR (neat) νmax: 3347, 3273, 3104, 2920, 1607, 1586, 1527, 1516, 1348, 1314, 1166, 1148, 933, 855, 742 cm−1. Mp 223 °C. 1H-NMR (CDCl3, 500 MHz): 8.46 (d, J = 8.9 Hz, 2H), 8.10 (d, J = 9.0 Hz, 2H), 7.57–7.55 (m, 2H), 7.43–7.41 (m, 3H), 7.38 (d, J = 8.1 Hz, 1H), 7.26 (t, J = 7.4 Hz, 1H), 7.14 (d, J = 7.6 Hz, 1H), 7.07 (t, J = 7.8 Hz, 1H), 5.07 (s, 1H), 4.78 (s, 1H), 4.29 (d, J = 14.3 Hz, 1H), 4.21 (d, J = 14.1 Hz, 1H), 3.84–3.79 (m, 1H), 3.71–3.66 (m, 1H), 2.11–2.06 (m, 1H), 1.93–1.88 (m, 1H). 13C-NMR (CDCl3, 75 MHz): 182.6 (Cq), 154.0 (Cq), 150.5 (Cq), 143.7 (Cq), 142.9 (Cq), 137.5 (Cq), 134.4 (2CH), 129.6 (CH), 129.5 (2CH), 129.0 (2CH), 128.7 (Cq), 128.0 (Cq), 124.8 (CH), 124.7 (2CH), 122.3 (CH), 120.4 (CH), 116.0 (CH2), 62.6 (Cq), 50.2 (CH2), 42.5 (CH2), 33.9 (CH2). HRMS (ESI): calcd. C25H22N3O4S2 [M + H]+ 492.1046, found 492.1052.
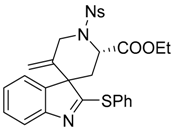
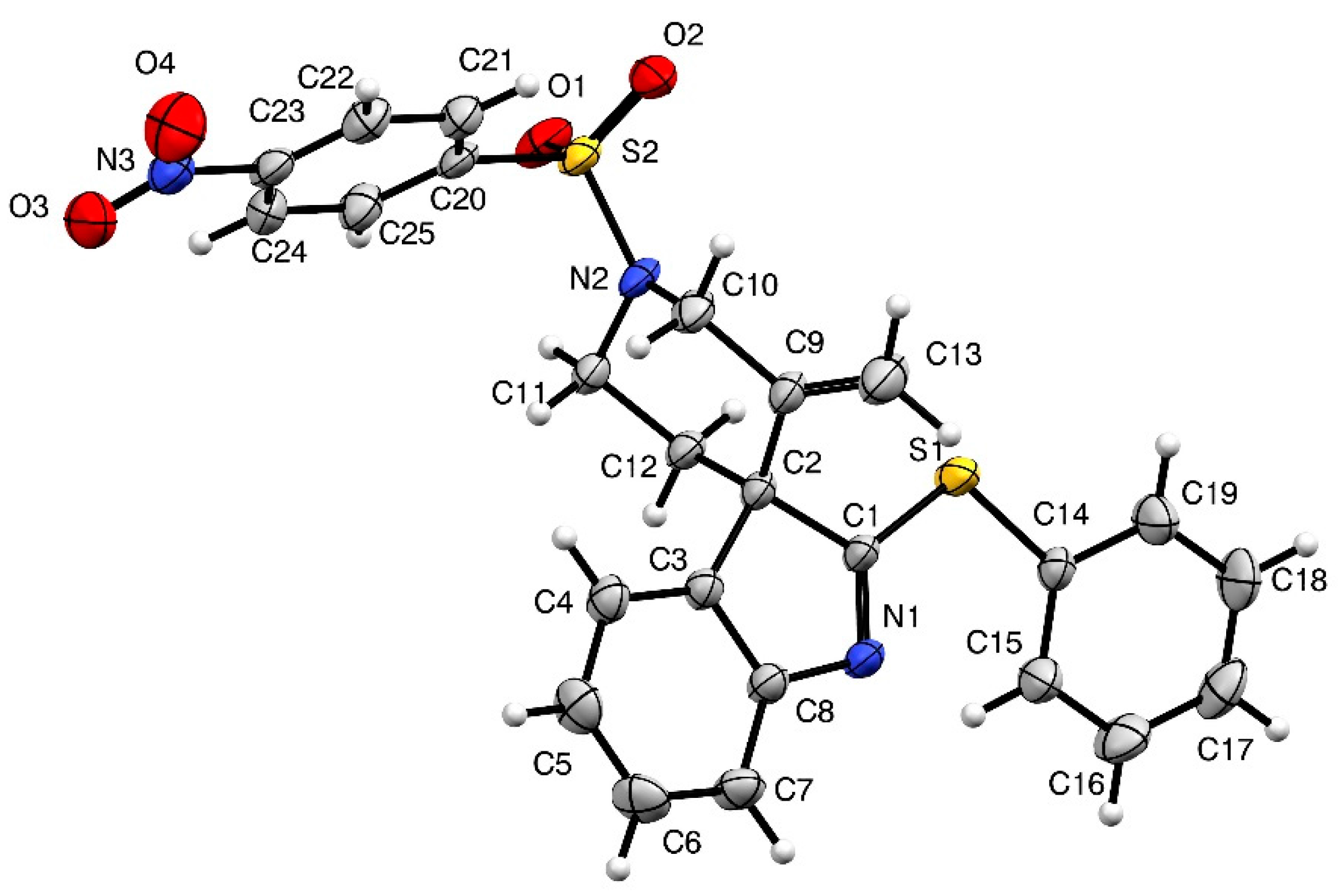
Ethyl-(2′S,3S)-5′-methylene-1′-[(4-nitrophenyl)sulfonyl]-2-(phenylthio)spiro[indole-3,4′-piperidine]-2′-carboxylate (4b)
Ethyl-(S)-2-{[4-nitro-N-(prop-2-yn-1-yl)phenyl]sulfonamide}-3-[2-(phenylthio)-1H-indol-3-yl]propanoate (7b) (56.4 mg, 0.1 mmol, 1 equiv.) was pourred in a shlenck tube filled with argon, dry toluene (1 mL) and PPh3AuNTf2 (3.7 mg, 0.005 mmol, 5 mol %) were added, and the solution was stirred at 70 °C for 15 h. Then the solvent was evaporated and the crude mixture was purified over silica gel column chromatography (10 to 30% EtOAc:Heptane), furnishing the pure product 4b as a yellowish solid (53.7 mg, 0.95 mmol, 95%). A 88/12 mixture of diastereoisomers was obtained and was characterized without further separation. For 1H-NMR only the most abundant diastereomer is described.
IR (neat) νmax: 3105, 3065, 2983, 1740, 1606, 1529, 1454, 1348, 1310, 1164, 1023, 909, 732 cm−1. Mp 82–83 °C. 1H-NMR of major diastereoisomer (CDCl3, 500 MHz): 8.41 (d, J = 8.9 Hz, 2H), 8.15 (d, J = 8.9 Hz, 2H), 7.62–7.60 (m, 2H), 7.45–7.41 (m, 4H), 7.29 (td, J = 7.5 and 1.2 Hz, 1H), 7.18–7.16 (m, 1H), 7.09 (td, J = 7.4 and 0.8 Hz, 1H), 5.12 (s, 1H), 4.83 (dd, J = 11.1 and 5.9 Hz, 1H), 4.60 (s, 1H), 4.47 (d, J = 15.3 Hz, 1H), 4.28 (dt, J = 15.2 and 1.8 Hz, 1H), 4.19-4.07 (m, 2H), 2.63 (dd, J = 14.2 and 11.0 Hz, 1H), 2.08 (dd, J = 14.2 and 6.0 Hz, 1H), 1.24 (t, J = 7.2 Hz, 3H). 13C-NMR of major diastereoisomer (CDCl3, 125 MHz): 183.6 (Cq), 171.0 (Cq), 154.4 (Cq), 150.3 (Cq), 145.6 (Cq), 144.0 (Cq), 138.9 (Cq), 134.4 (2CH), 129.6 (CH), 129.6 (2CH), 129.1 (2CH), 128.8 (CH), 127.7 (Cq), 125.4 (CH), 124.4 (2CH), 122.4 (CH), 120.3 (CH), 115.7 (CH2), 62.3 (CH2), 61.3 (Cq), 53.9 (CH), 47.6 (CH2), 35.1 (CH2), 14.2 (CH3). 13C-NMR of minor diastereoisomer (CDCl3, 125 MHz): 183.3 (Cq), 170.8 (Cq), 153.5 (Cq), 145.6 (Cq), 143.5 (Cq), 137.3 (Cq), 134.7 (Cq), 134.4 (2CH), 129.0 (2CH), 128.7 (CH), 127.9 (Cq), 124.7 (CH), 124.4 (2CH), 121.9 (CH), 111.5 (CH2), 62.3 (CH2), 61.9 (Cq), 53.7 (CH), 47.5 (CH2), 35.9 (CH2), 14.0 (CH3). HRMS (ESI): calcd. for C28H26N3O6S2 [M + H]+ 564.1258, found 464.1254.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}