N-Vinylation of Imidazole and Benzimidazole with a Paramagnetic Vinyl Bromide

Abstract
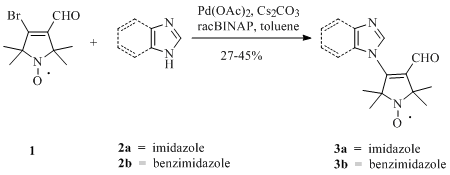
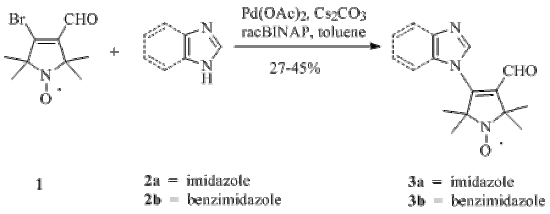{kind=link}
{kind=link}
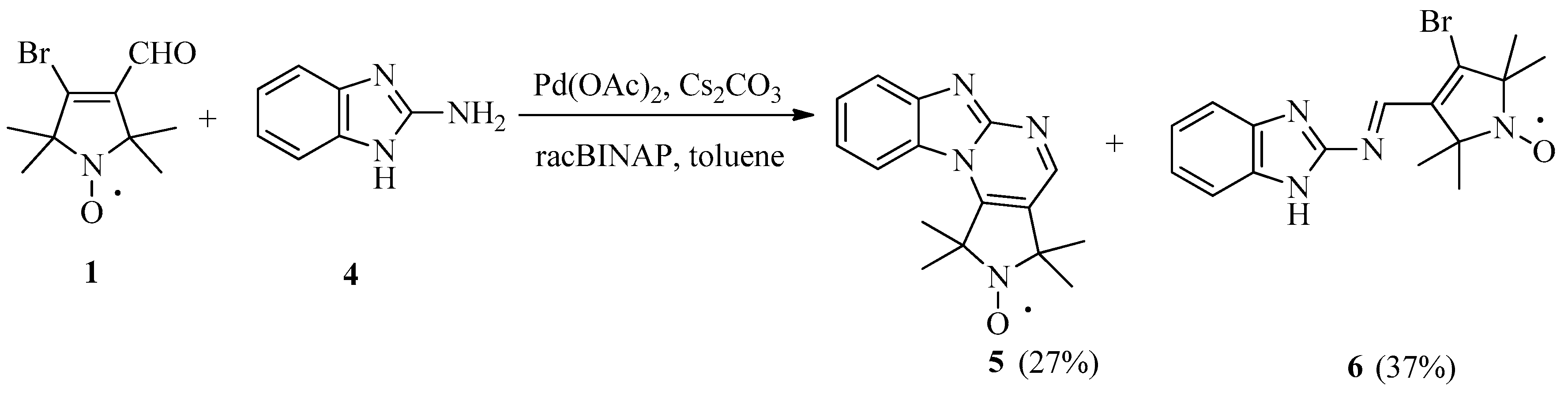{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
Pd-Catalyzed N-Vinylation, General Procedure (3a, 3b, 5, 6)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wertz, S.; Studer, A. Nitroxide-catalyzed transition-metal-free aerobic oxidation processes. Green Chem. 2013, 15, 3116–3134. [Google Scholar] [CrossRef]
- Haugland, M.M.; Lovett, J.E.; Anderson, E.A. Advances in the synthesis of nitroxide for use in biomolecule spin labeling. Chem. Soc. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, M.; Gwozdzinski, K. Nitroxides as Antioxidants and Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 2490. [Google Scholar] [CrossRef] [PubMed]
- Kálai, T.; Kuppusamy, M.L.; Madan, E.; Bognár, B.; Balog, M.; Jekő, J.; Kuppusamy, P.; Hideg, K. Synthesis and biological evaluation of new paramagnetic curcumin analogues. Med. Chem. 2017, 13, 761–772. [Google Scholar]
- Gigmes, D. Nitroxide Mediated Polymerization: From Fundamentals to Applications in Materials Science; Royal Society of Chemistry: London, UK, 2015. [Google Scholar]
- Winsberg, J.; Hagemann, T.; Janoschka, T.; Hager, M.D.; Schubert, U.S. Redox-Flow Batteries: From Metals to Organic Redox-Active Materials. Angew. Chem. Int. Ed. 2017, 56, 686–711. [Google Scholar] [CrossRef] [PubMed]
- Lussini, C.V.; Colwell, J.M.; Fairfull-Smith, K.E.; Bottle, S.E. Profluorescent nitroxide sensors for monitoring photo-induced degradation in polymer films. Sens. Actuators B 2017, 241, 199–209. [Google Scholar] [CrossRef]
- Hilt, S.; Tang, T.; Walton, J.H.; Budamagunta, M.; Maetawa, I.; Kálai, T.; Hideg, K.; Singh, V.; Wulff, H.; Gong, Q.; et al. A Metal-Free Method for Producing MRI Contrast at Amyloid-β. J. Alzheimer Dis. 2017, 55, 1667–1681. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, V.V.; Bobko, A.A.; Tseytlin, M.; Driesschaert, B. Exchange Phenomena in the Electron Paramagnetic Resonance Spectra of the Nitroxyl and Trityl Radicals: Multifunctional Spectroscopy and Imaging of Local Chemical Microenvironment. Anal. Chem. 2017, 89, 4758–4771. [Google Scholar] [CrossRef] [PubMed]
- Kálai, T.; Balog, M.; Jekő, J.; Hubbell, W.L.; Hideg, K. Palladium Catalysed Coupling Reactions of Paramagnetic Vinyl Halides. Synthesis 2002, 34, 2365–2372. [Google Scholar]
- Kokorin, A.I.; Zaripov, R.B.; Gromov, O.I.; Sukhanov, A.A.; Kálai, T.; Lamperth, É.; Hideg, K. Spin Density Distribution in a Nitroxide Biradical Containing 13C-Enriched Acetylene Groups in the Bridge: DFT Calculations and EPR Investigation. Appl. Magn. Reson. 2016, 47, 1057–1067. [Google Scholar] [CrossRef][Green Version]
- Keddie, D.J.; Johnson, T.E.; Arnold, D.P.; Bottle, S.E. Synthesis of profluorescent isoindoline nitroxides via palladium-catalysed Heck alkenylation. Org. Biomol. Chem. 2005, 3, 2593–2598. [Google Scholar] [CrossRef] [PubMed]
- Kálai, T.; Bognár, B.; Zsolnai, D.; Berente, Z.; Hideg, K. Synthesis of Nitroxide annulated carbocycles and heterocycles. Synthesis 2012, 44, 3655–3660. [Google Scholar] [CrossRef]
- Úr, G.; Kálai, T.; Hideg, K. Facile syntheses of 3,4-disubstituted pyrroline nitroxides and their further synthetic applications. Tetrahedron Lett. 2016, 57, 778–780. [Google Scholar] [CrossRef]
- Úr, G.; Fekete, G.G.; Jekő, J.; Hideg, K.; Kálai, T. Palladium- and/or Copper-Catalyzed Cross-Coupling Reactions of Paramagnetic Vinyl Bromides and Iodides. Synthesis 2017, 49, 3740–3748. [Google Scholar]
- Zhdanov, R.I. Nitroxyl Radicals and Non-Radical Reactions of Free Radicals. In Bioactive Spin Labels; Zhdanov, R.I., Ed.; Springer: Berlin, Germany, 1992; pp. 23–82. [Google Scholar]
- Hesse, S.; Kirsch, G. Buchwald-Hartwig amination of β-chloroacroleins by Lactams and Heteroarylamines. Synthesis 2007, 39, 1571–1575. [Google Scholar] [CrossRef]
- Mao, J.; Hua, Q.; Guo, J.; Shi, D.; Ji, S. CuI-Catalyzed Cross-coupling Reactions of β-Vinyl Bromides with Nitrogen–Containing Heterocycles. Synlett 2008, 19, 2011–2016. [Google Scholar] [CrossRef]
- Ho, S.L.; Dao, P.D.Q. Microwave-Assisted Synthesis of Benzo[4,5]imidazo[1,2-a]pyrimidines from β-bromo-α,β-unsaturated aldehydes and 2-aminobenzimidazoles. Synlett 2017, 28, 1811–1815. [Google Scholar]
- Hankovszky, H.O.; Hideg, K.; Lex, L. Nitroxyls; VII1. Synthesis and Reactions of Highly Reactive 1-Oxyl-2,2,5,5-tetramethyl-2,5-dihydropyrrole-3-ylmethyl Sulfonates. Synthesis 1980, 12, 914–916. [Google Scholar] [CrossRef]
- Berliner, L.J.; Grünwald, J.; Hankovszky, H.O.; Hideg, K. A Novel Reversible Thiol-Specific Spin Label: Papain Active Site Labeling and Inhibition. Anal. Biochem. 1982, 119, 450–455. [Google Scholar] [CrossRef]
- Sár, P.C.; Kálai, T.; Bárácz, M.N.; Jerkovich, G.; Hideg, K. Selective Reduction of Nitrones and Nitroxides to Functionalized Secondary Amines. Synth. Commun. 1995, 25, 2929–2940. [Google Scholar] [CrossRef]
- Shelke, S.A.; Sigurdsson, S.T. Site Directed Spin Labeling for EPR Studies of Nucleic Acids. In Modified Nucleic Acids; Nakatani, K., Tor, Y., Eds.; Springer: Basel, Switzerland, 2016; pp. 159–187. [Google Scholar]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Úr, G.; Gulyás Fekete, G.; Hideg, K.; Kálai, T. N-Vinylation of Imidazole and Benzimidazole with a Paramagnetic Vinyl Bromide. Molbank 2018, 2018, M980. https://doi.org/10.3390/M980
Úr G, Gulyás Fekete G, Hideg K, Kálai T. N-Vinylation of Imidazole and Benzimidazole with a Paramagnetic Vinyl Bromide. Molbank. 2018; 2018(1):M980. https://doi.org/10.3390/M980
Chicago/Turabian StyleÚr, Györgyi, Gergely Gulyás Fekete, Kálmán Hideg, and Tamás Kálai. 2018. "N-Vinylation of Imidazole and Benzimidazole with a Paramagnetic Vinyl Bromide" Molbank 2018, no. 1: M980. https://doi.org/10.3390/M980
APA StyleÚr, G., Gulyás Fekete, G., Hideg, K., & Kálai, T. (2018). N-Vinylation of Imidazole and Benzimidazole with a Paramagnetic Vinyl Bromide. Molbank, 2018(1), M980. https://doi.org/10.3390/M980