(S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione—Synthesis and Crystallographic Studies

 ,
, 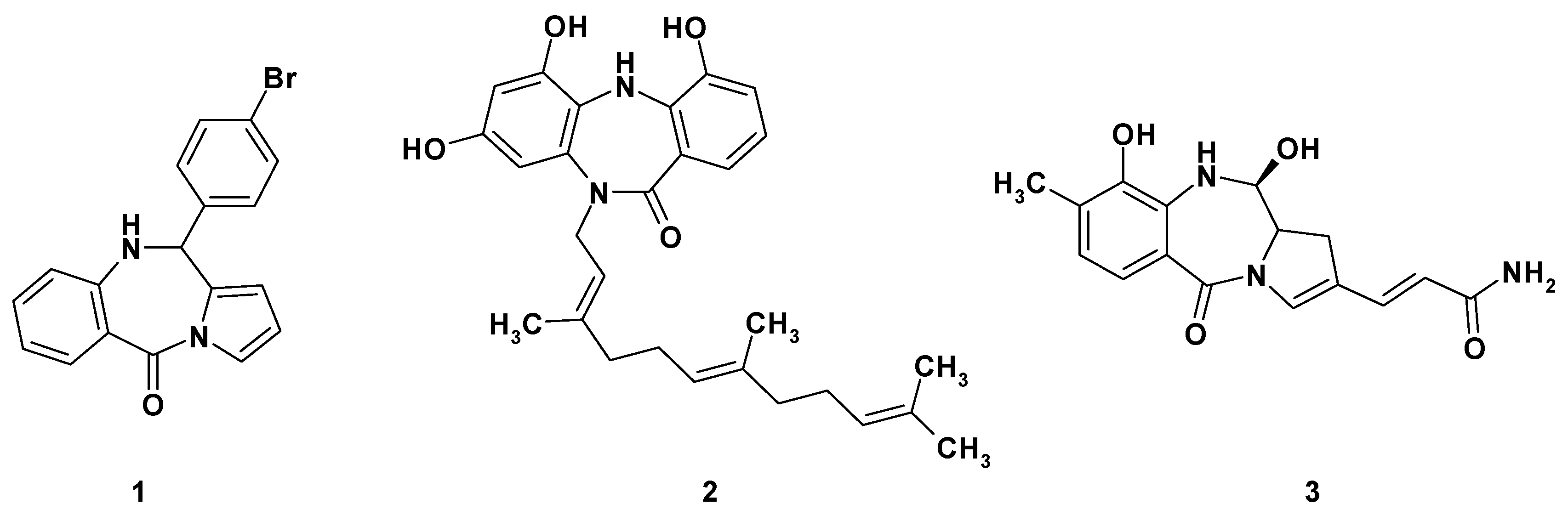{kind=link}
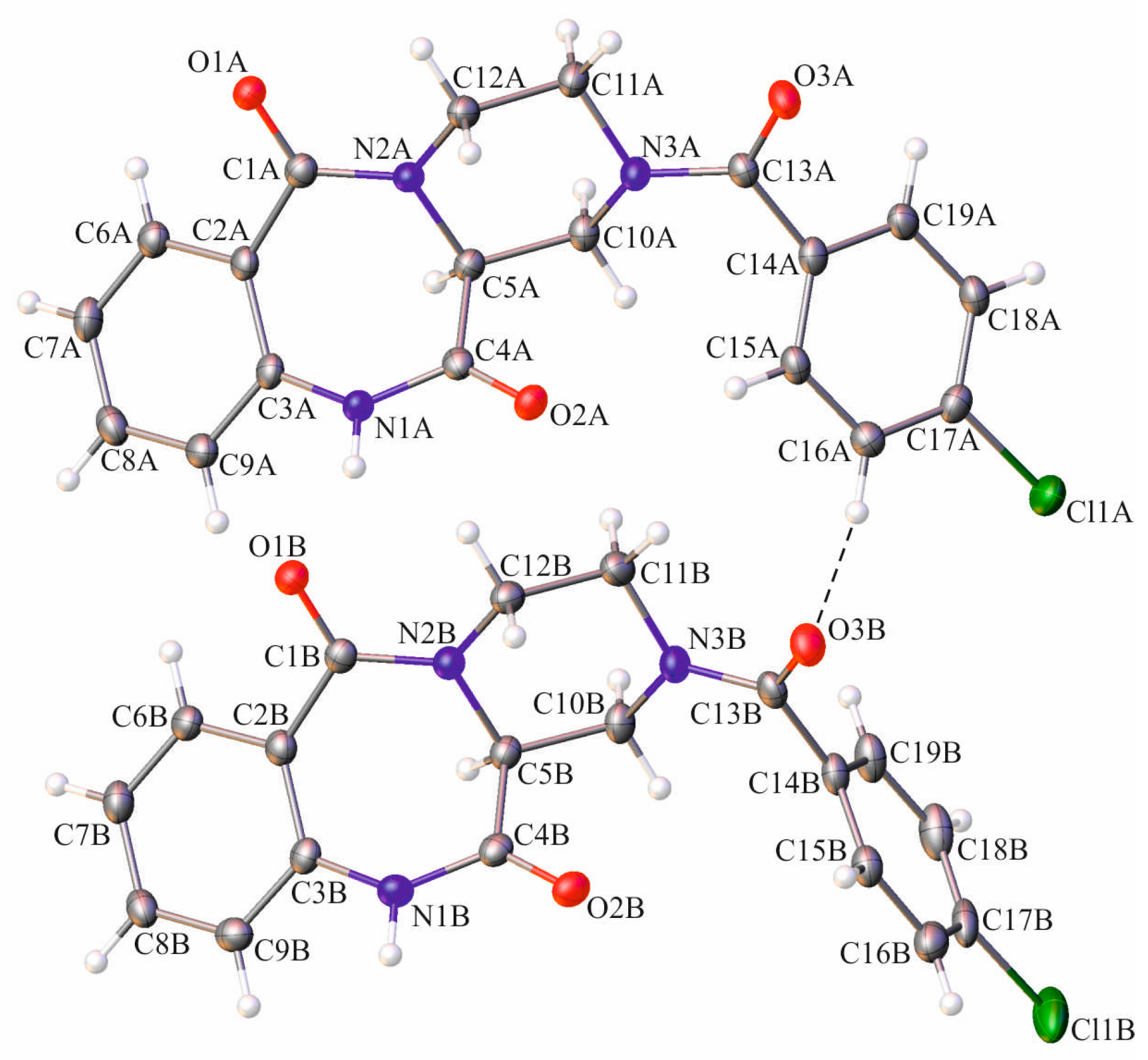{kind=link}
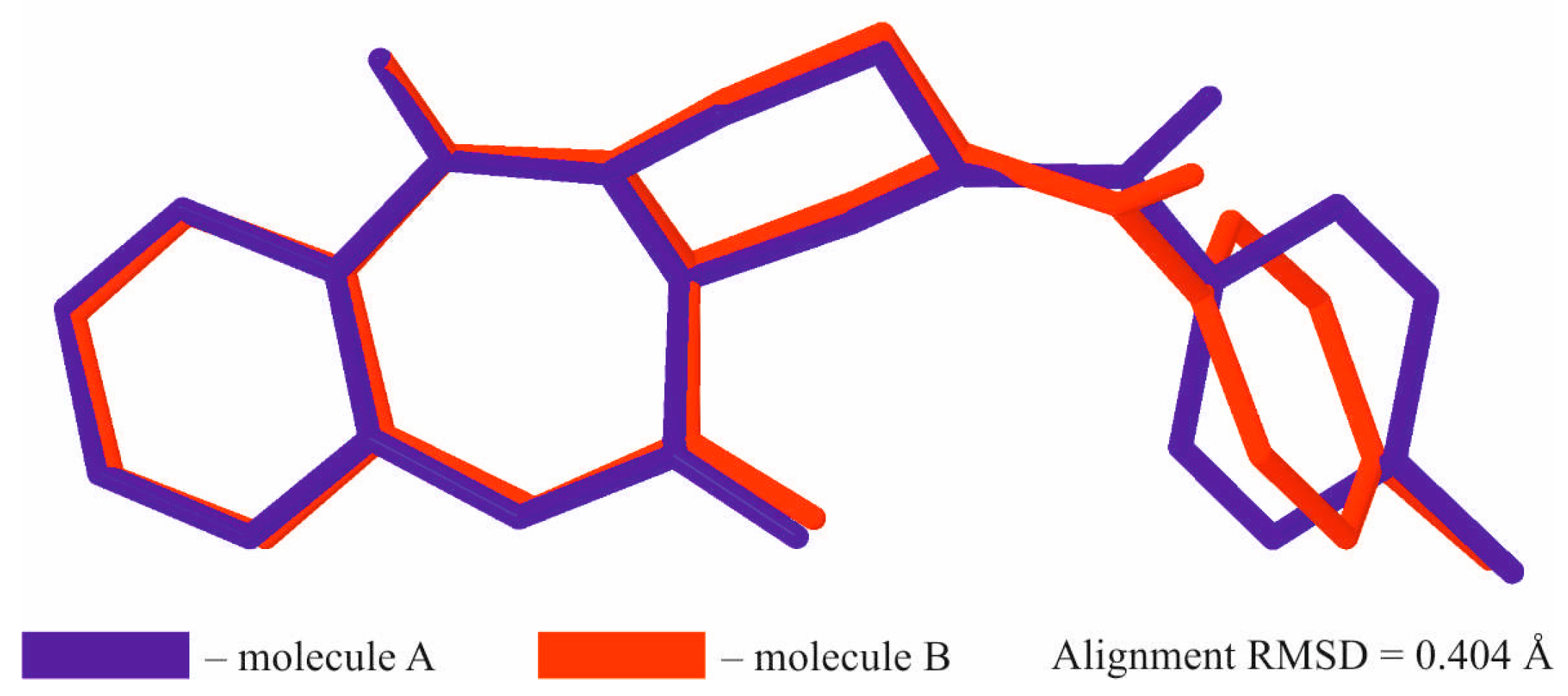{kind=link}
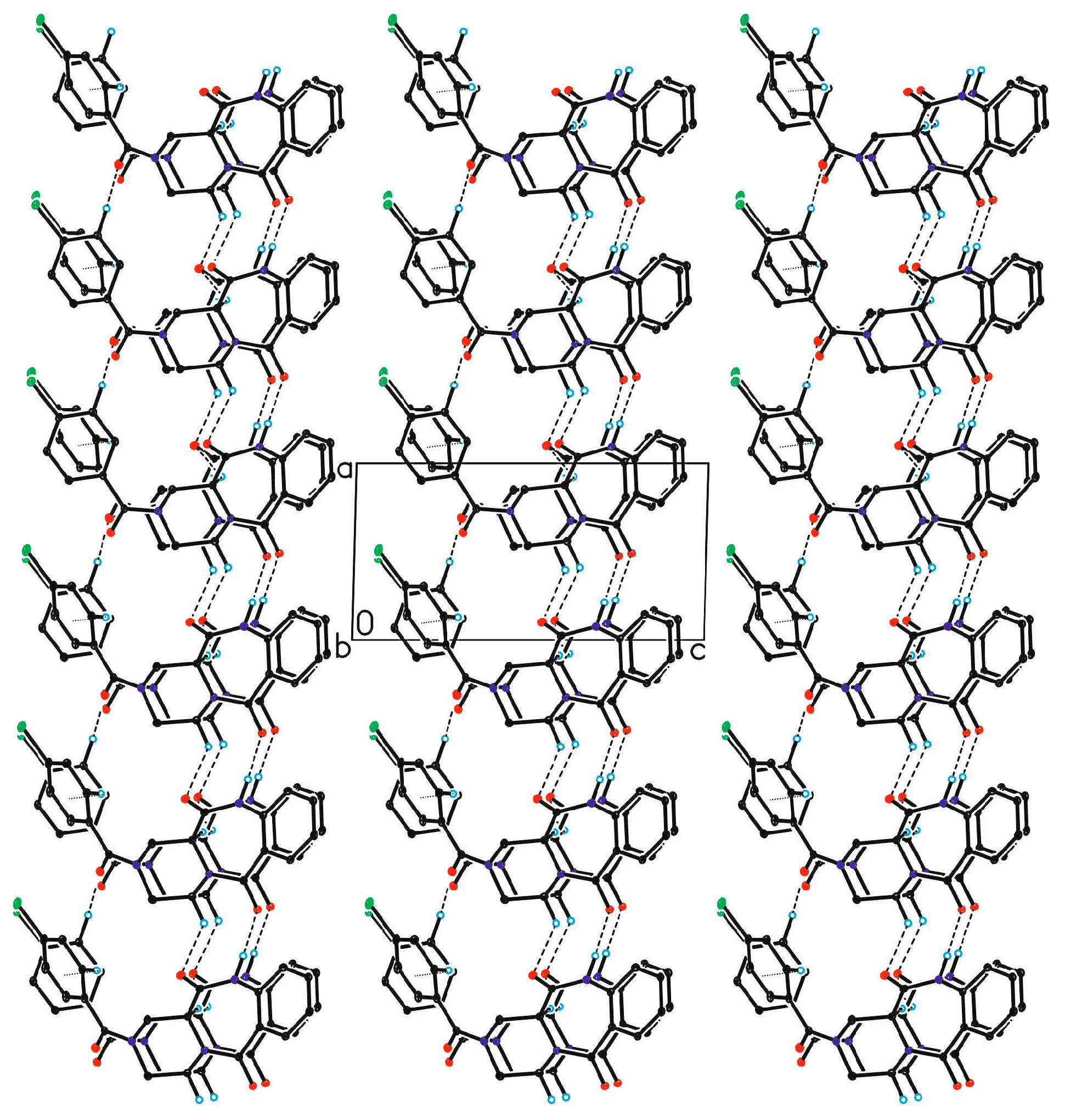{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
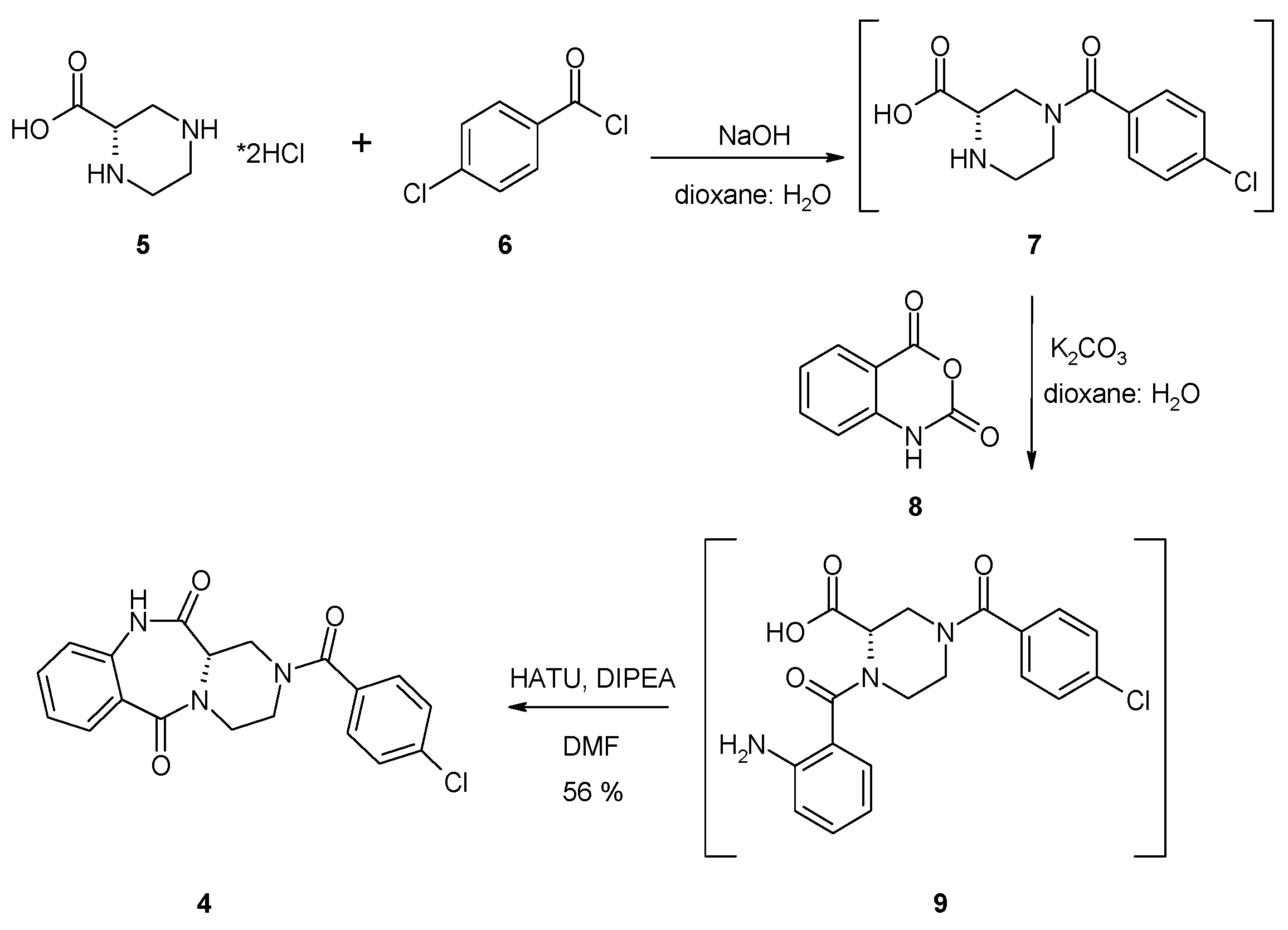
Synthesis of the (S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione (4)
Supplementary Materials
Acknowledgments
Author Contributions
References
- Shameem, M.; Kumar, R.; Krishna, S.; Kumar, C.; Siddiqui, M.I.; Kundu, B.; Banerjee, D. Synthetic modified pyrrolo[1,4]benzodiazepine molecules demonstrate selective anticancer activity by targeting the human ligase 1 enzyme: An in silico and in vitro mechanistic study. Chem. Biol. Interact. 2015, 237, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Rambabu, V.; Vijayakumar, S. In vitro cytotoxicity perspective of deazepinomycin (ECO-4601) on human hepatoma cell line (HEPG2). Biomed. Aging Pathol. 2014, 4, 65–70. [Google Scholar] [CrossRef]
- Cipolla, L.; Araujo, A.C.; Airoldi, C.; Bini, D. Pyrrolo[2,1-c]benzodiazepine as a scaffold for the design and synthesis of antitumor drugs. Anti-Cancer Med. Chem. 2009, 9, 1–31. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Makowska, M.; Sekuła, J.; Tomczyk, E.; Zalewska, E.; Nasulewicz-Goldeman, A.; Wietrzyk, J. Bicyclic cytarabine analogues: Synthesis and investigation of antitumor properties of novel, 6-aryl- and 6-alkyl-3H-pyrrolo[2,3-d]pyrimidin-2(7H)-one arabinosides. Tetrahedron 2015, 71, 8454–8461. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Tomczyk, E.; Makowska, M.; Nasulewicz-Goldeman, A.; Gajda, R.; Woźniak, K.; Wietrzyk, J. The synthesis and investigation of anti-tumor properties of novel, bicyclic furopyrimidine, pyrrolopyrimidine and pyrimidopyridazine nucleoside analogues. Synthesis 2016, 48, 566–572. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Bazlekova, M.; Bagiński, M.; Wójcik, J.; Winczura, A.; Miazga, A.; Gajda, R.; Woźniak, K.; Tudek, B. A mild and efficient approach to 6H-oxazolo[3,2-f]pyrimidine-5,7-dione scaffold via unexpected rearrangement of 2,3-dihydropyrimido[6,1-b][1,5,3]dioxazepine-7,9(5H,8H)-diones: A synthesis, crystallographic studies and cytotoxic activity screening. Tetrahedron Lett. 2016, 57, 743–746. [Google Scholar] [CrossRef]
- Mieczkowski, A.; Jurczak, J. A traceless solid-supported synthesis of novel pyrazinediazepinedione derivatives. Tetrahedron 2010, 66, 2514–2519. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- CrysAlis CCD and CrysAlis RED, Version 1.171.32.15; Oxford Diffraction Ltd.: Yarnton, UK, 2008.
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mieczkowski, A.; Trzybiński, D.; Wilczek, M.; Psurski, M.; Bagiński, M.; Bieszczad, B.; Mroczkowska, M.; Woźniak, K. (S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione—Synthesis and Crystallographic Studies. Molbank 2017, 2017, M964. https://doi.org/10.3390/M964
Mieczkowski A, Trzybiński D, Wilczek M, Psurski M, Bagiński M, Bieszczad B, Mroczkowska M, Woźniak K. (S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione—Synthesis and Crystallographic Studies. Molbank. 2017; 2017(4):M964. https://doi.org/10.3390/M964
Chicago/Turabian StyleMieczkowski, Adam, Damian Trzybiński, Marcin Wilczek, Mateusz Psurski, Maciej Bagiński, Bartosz Bieszczad, Magdalena Mroczkowska, and Krzysztof Woźniak. 2017. "(S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione—Synthesis and Crystallographic Studies" Molbank 2017, no. 4: M964. https://doi.org/10.3390/M964
APA StyleMieczkowski, A., Trzybiński, D., Wilczek, M., Psurski, M., Bagiński, M., Bieszczad, B., Mroczkowska, M., & Woźniak, K. (2017). (S)-2-(4-Chlorobenzoyl)-1,2,3,4-tetrahydrobenzo[e]pyrazino[1,2-a][1,4]diazepine-6,12(11H,12aH)-dione—Synthesis and Crystallographic Studies. Molbank, 2017(4), M964. https://doi.org/10.3390/M964