Synthesis and Detailed Spectroscopic Characterization of Two Novel N-(3-Acetyl-2-thienyl)acetamides

{kind=link}
Abstract
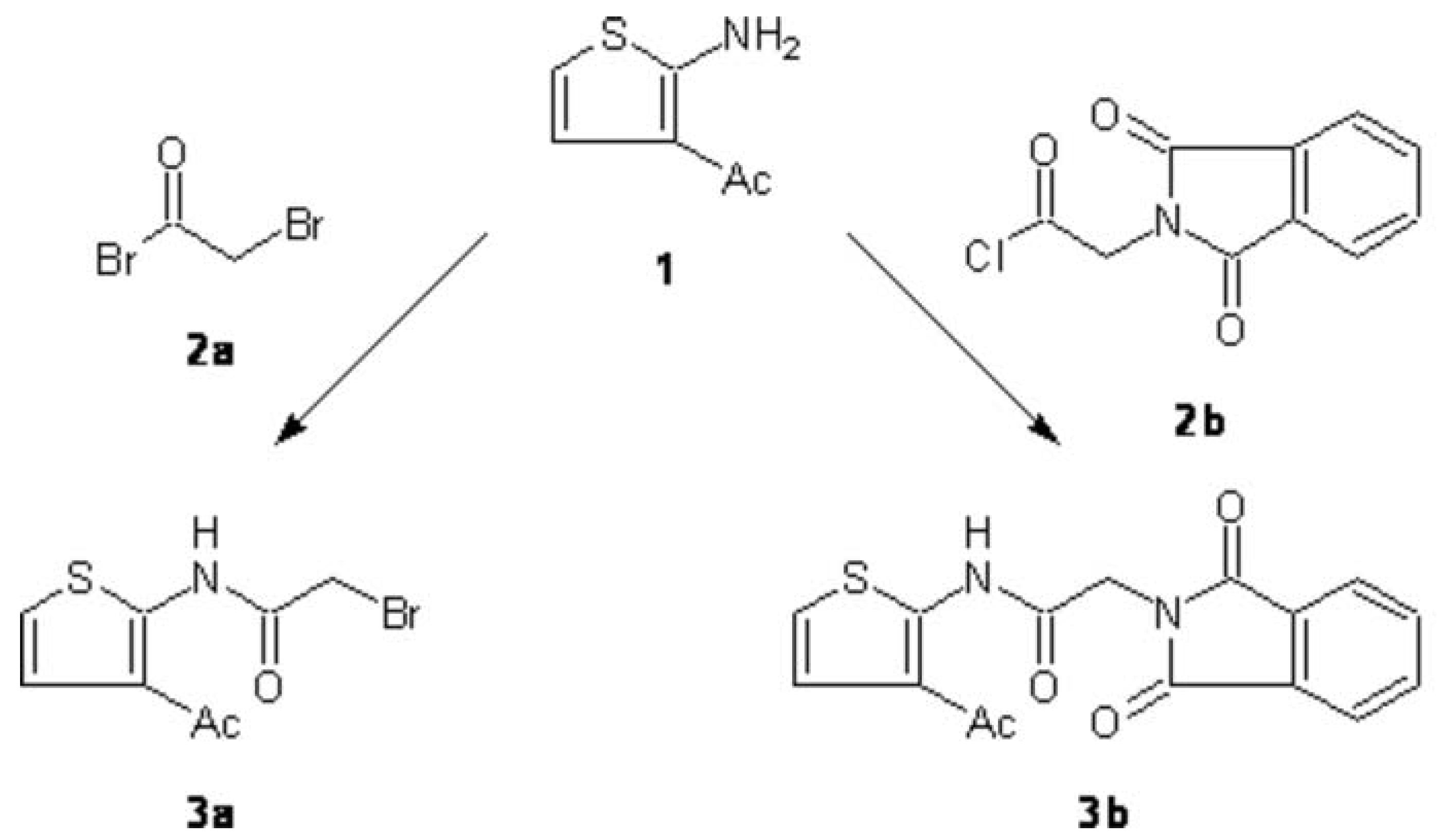
N-(3-Acetyl-2-thienyl)-2-bromoacetamide (3a):
N-(3-Acetyl-2-thienyl)-2-(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)acetamide (3b):
Supplementary materials
Supplementary File 1Supplementary File 2Supplementary File 3Supplementary File 4Supplementary File 5Supplementary File 6References and Notes
- Gewald, K. Chem. Ber. 1965, 98, 3571–3577.
- Eller, G. A.; Holzer, W. Molecules 2006, 11, 371–376. [PubMed]
- The spectrum was obtained on a Perkin-Elmer FTIR 1605 spectrophotometer.
- The spectrum was obtained on a Varian UnityPlus 300 spectrometer (299.95 MHz for 1H, 75.43 MHz for 13C) at 28 °C. The center of the solvent signal was used as an internal standard which was related to TMS with δ 2.49 ppm (1H NMR) and δ 39.5 ppm (13C NMR).
- The spectrum was obtained on a Bruker Avance 500 spectrometer (500.13 MHz for 1H, 125.77 MHz for 13C) at 294 K. The center of the solvent signal was used as an internal standard which was related to TMS with δ 7.26 ppm (1H NMR) and δ 77.0 ppm (13C NMR).
- The spectrum was obtained on a Bruker Avance 500 spectrometer (50.68 MHz for 15N) and was referenced against neat, external nitromethane (coaxial capillary).
- The spectrum was obtained on a Shimadzu QP 1000 instrument (EI, 70eV).
- Usifoh, C. O.; Lambert, D. M.; Wouters, J.; Scriba, G. K. E. Arch. Pharm. (Weinheim, Ger.) 2001, 334, 323–331.
© 2006 MDPI. All rights reserved.
Share and Cite
Eller, G.A.; Holzer, W. Synthesis and Detailed Spectroscopic Characterization of Two Novel N-(3-Acetyl-2-thienyl)acetamides. Molbank 2006, 2006, M520. https://doi.org/10.3390/M520
Eller GA, Holzer W. Synthesis and Detailed Spectroscopic Characterization of Two Novel N-(3-Acetyl-2-thienyl)acetamides. Molbank. 2006; 2006(6):M520. https://doi.org/10.3390/M520
Chicago/Turabian StyleEller, Gernot A., and Wolfgang Holzer. 2006. "Synthesis and Detailed Spectroscopic Characterization of Two Novel N-(3-Acetyl-2-thienyl)acetamides" Molbank 2006, no. 6: M520. https://doi.org/10.3390/M520
APA StyleEller, G. A., & Holzer, W. (2006). Synthesis and Detailed Spectroscopic Characterization of Two Novel N-(3-Acetyl-2-thienyl)acetamides. Molbank, 2006(6), M520. https://doi.org/10.3390/M520