Recent Developments in Peptide-Based Nucleic Acid Delivery

Abstract
:1. Introduction
2. General Properties of CPPs
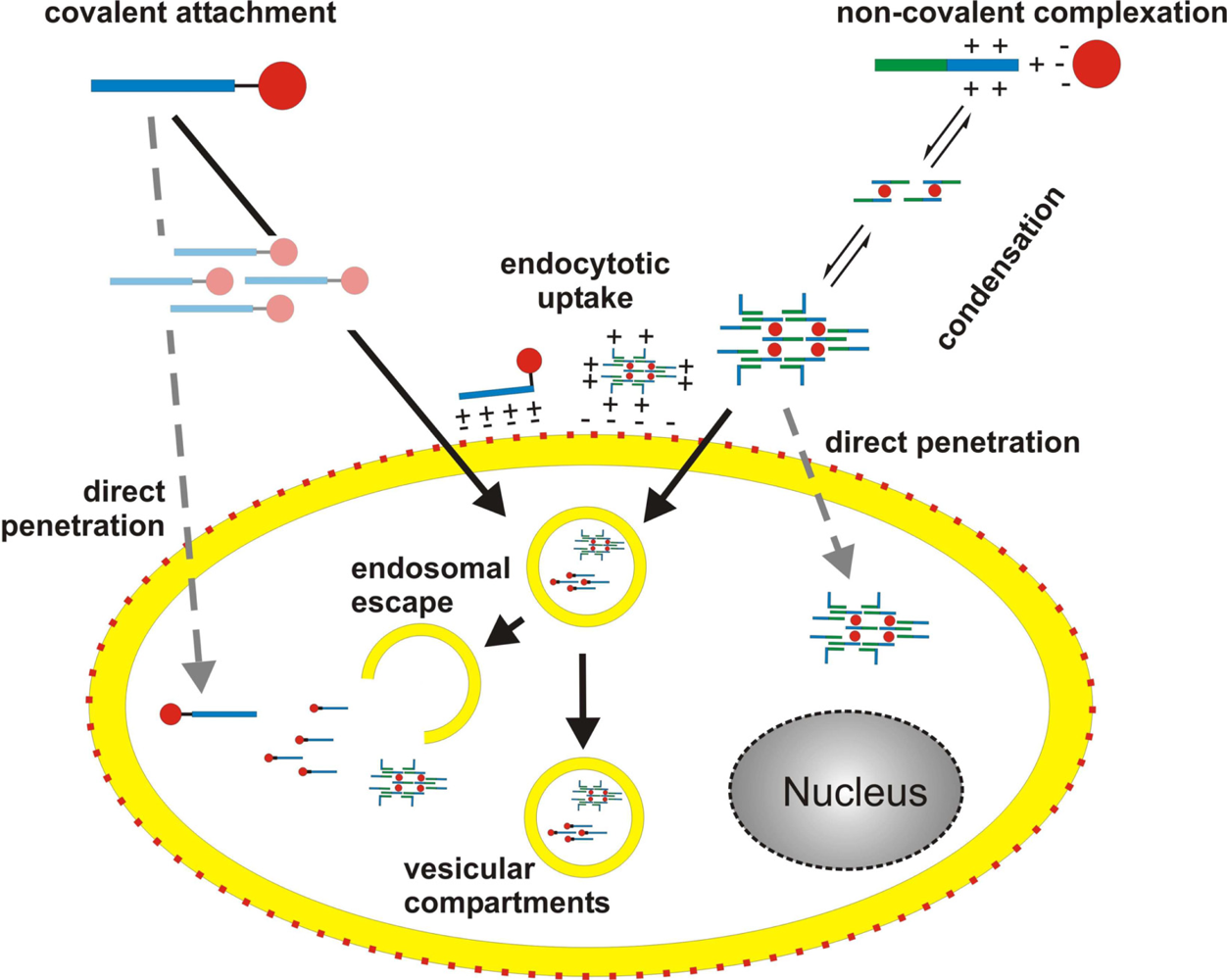
3. Mechanism of cellular translocation
4. Properties of selected CPPs
4.1. Classical CPPs
HIV-1 Tat and derivatives
Penetratin
Transportan and derivatives
Oligoarginines
Model amphipathic peptides
MPG and Pep families
4.2. New peptides and concepts
Human calcitonin and derivatives
ARF-protein derived peptide M918
Sweet arrow peptide
Dermaseptin derived peptide S413-PV
Prion protein derived peptides
Cell-penetrating pentapeptides (CPP5)
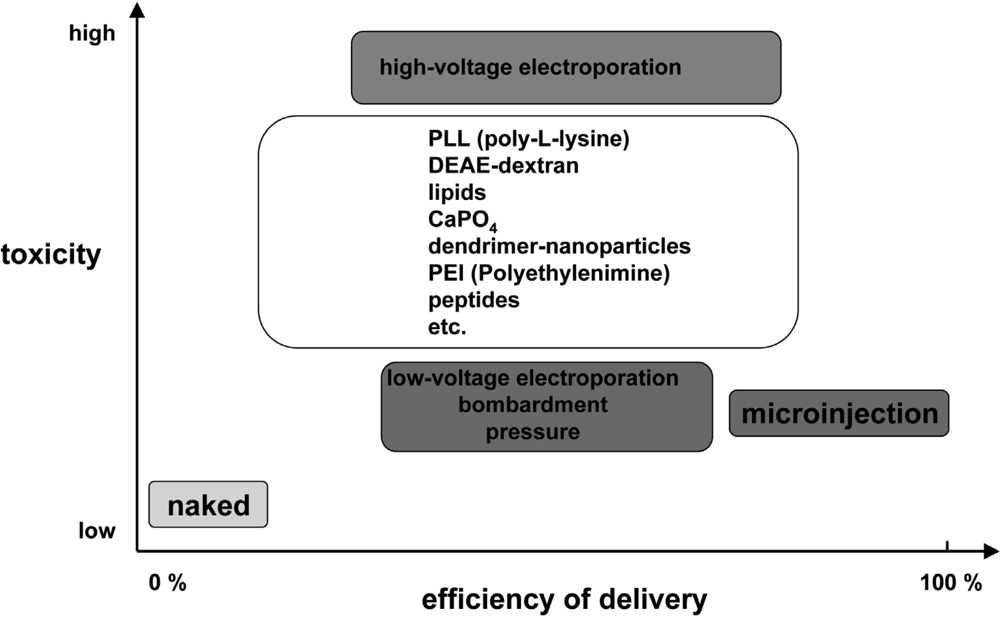
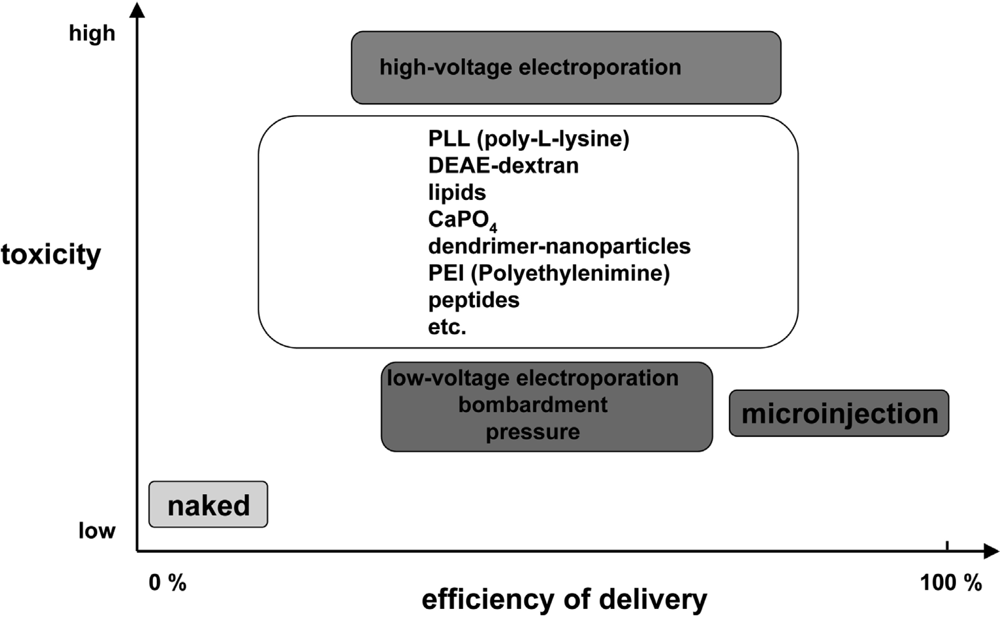
Polymers and complex systems
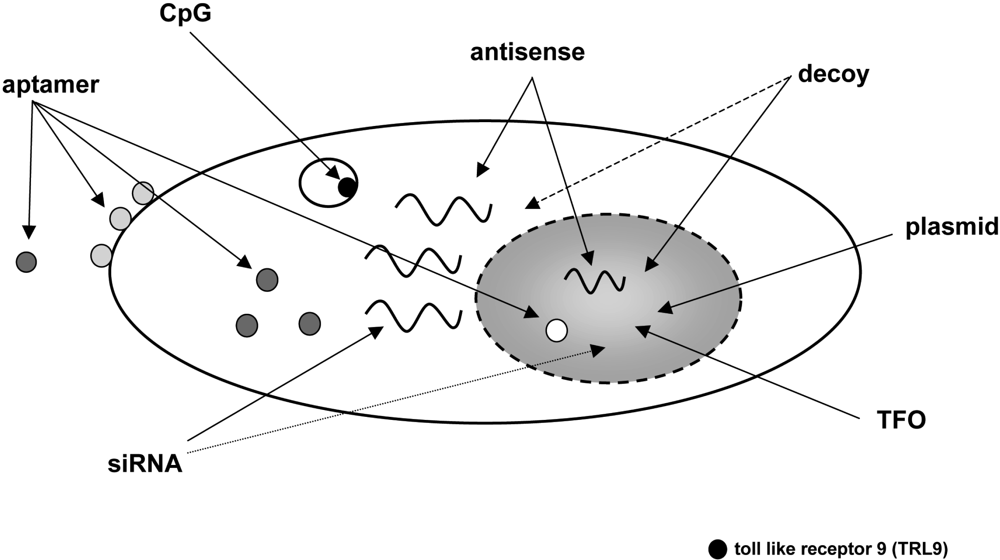
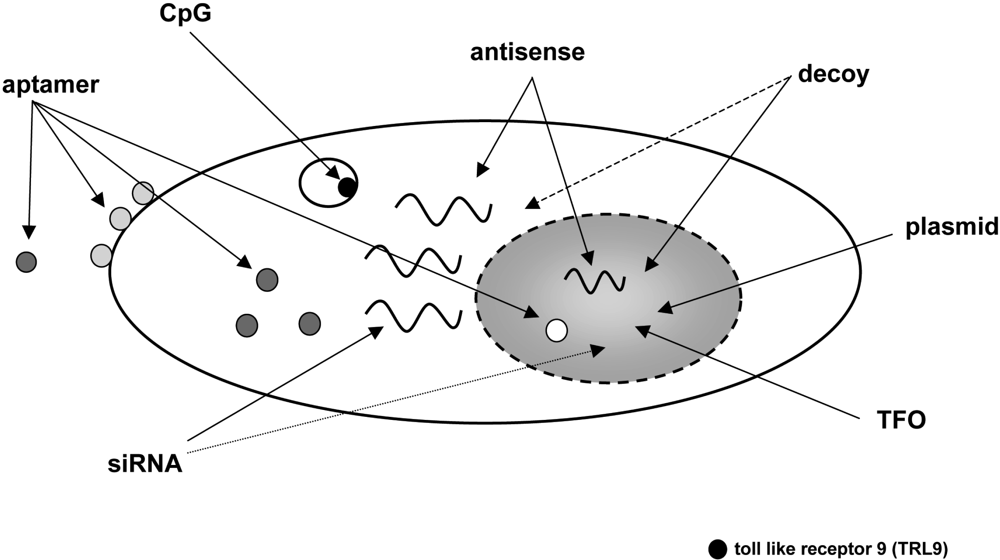
5. CPP-mediate delivery of nucleic acids
5.1. Plasmids
5.2. Antisense oligonucleotides
5.3. Transcription factor decoy oligonucleotides and triplex forming oligonucleotides
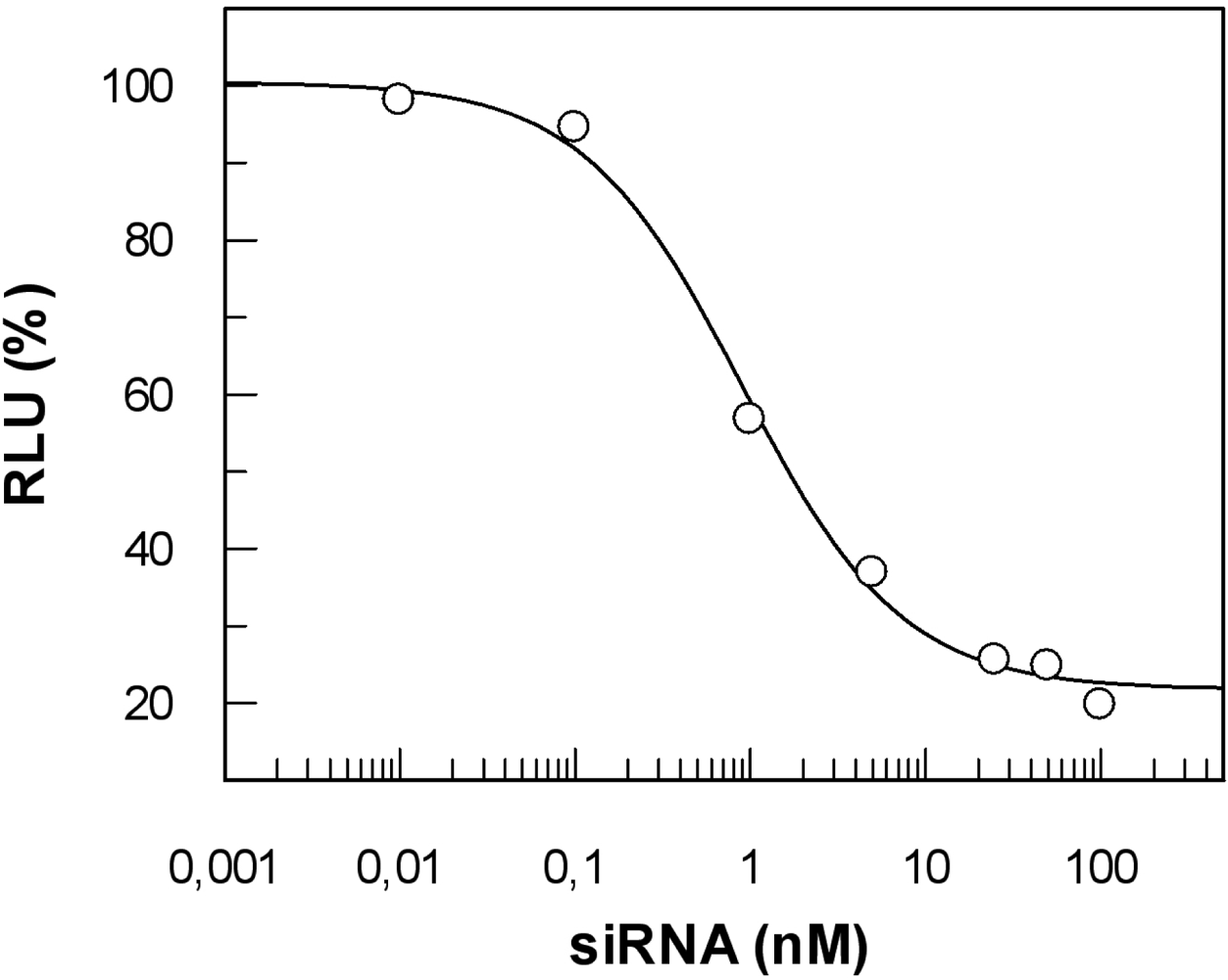
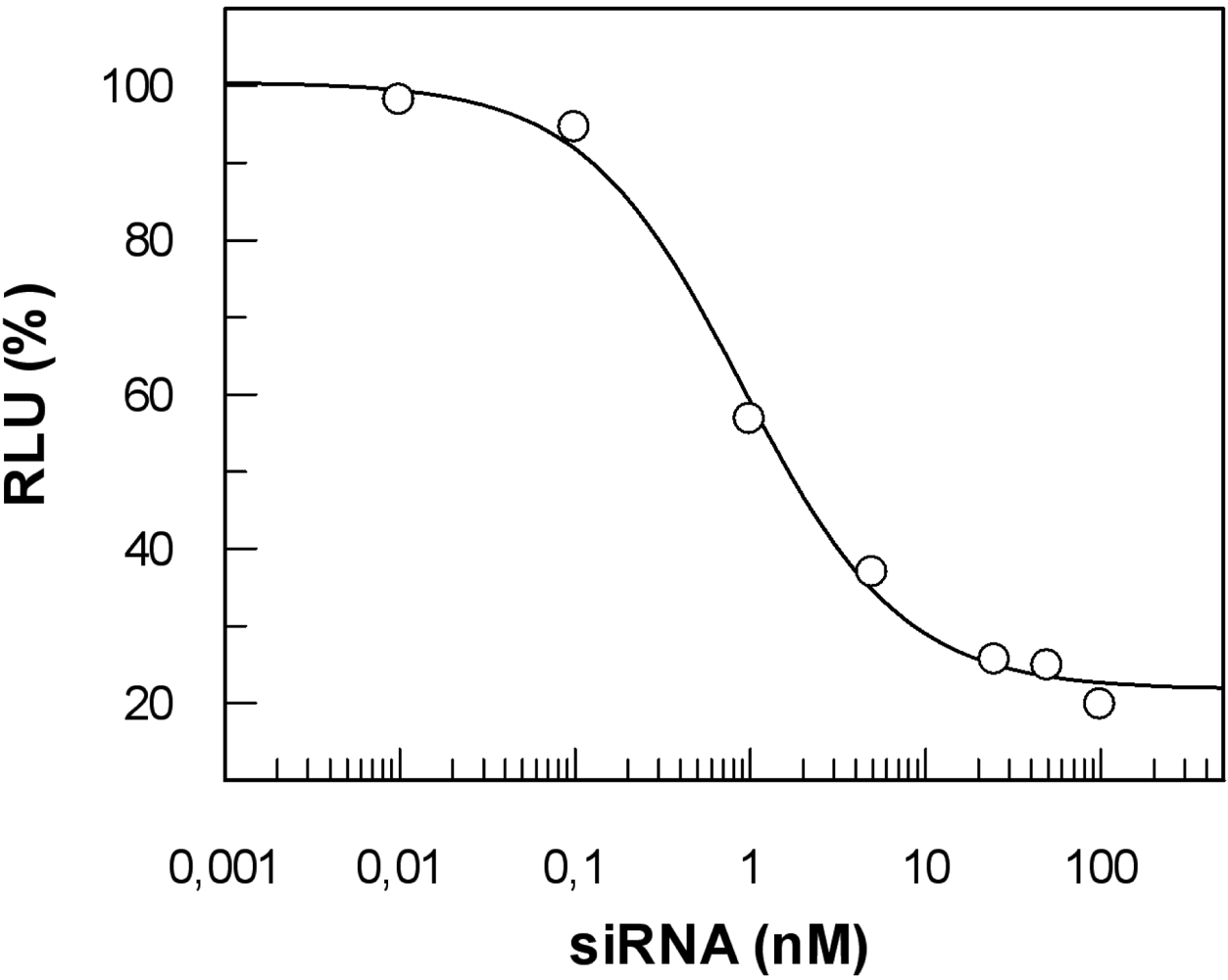
5.4. siRNA
6. Cellular uptake versus functional effects of the nucleic acid cargo
7. Conclusion
Acknowledgments
References and Notes
- Opalinska, JB; Gewirtz, AM. Nucleic-acid therapeutics: basic principles and recent applications. Nat Rev Drug Discov 2002, 1, 503–514. [Google Scholar]
- Eckstein, F. The versatility of oligonucleotides as potential therapeutics. Expert Opin Biol Ther 2007, 7, 1021–1034. [Google Scholar]
- Kootstra, NA; Verma, IM. Gene therapy with viral vectors. Annu Rev Pharmacol Toxicol 2003, 43, 413–439. [Google Scholar]
- Verma, IM; Weitzman, MD. Gene therapy: twenty-first century medicine. Annu Rev Biochem 2005, 74, 711–738. [Google Scholar]
- Raper, SE; Yudkoff, M; Chirmule, N; Gao, GP; Nunes, F; Haskal, ZJ; Furth, EE; Propert, KJ; Robinson, MB; Magosin, S; Simoes, H; Speicher, L; Hughes, J; Tazelaar, J; Wivel, NA; Wilson, JM; Batshaw, ML. A pilot study of in vivo liver-directed gene transfer with an adenoviral vector in partial ornithine transcarbamylase deficiency. Hum Gene Ther 2002, 13, 163–175. [Google Scholar]
- Hacein-Bey-Abina, S; Von Kalle, C; Schmidt, M; McCormack, MP; Wulffraat, N; Leboulch, P; Lim, A; Osborne, CS; Pawliuk, R; Morillon, E; Sorensen, R; Forster, A; Fraser, P; Cohen, JI; de Saint, BG; Alexander, I; Wintergerst, U; Frebourg, T; Aurias, A; Stoppa-Lyonnet, D; Romana, S; Radford-Weiss, I; Gross, F; Valensi, F; Delabesse, E; Macintyre, E; Sigaux, F; Soulier, J; Leiva, LE; Wissler, M; Prinz, C; Rabbitts, TH; Le Deist, F; Fischer, A; Cavazzana-Calvo, M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar]
- Raper, SE; Chirmule, N; Lee, FS; Wivel, NA; Bagg, A; Gao, GP; Wilson, JM; Batshaw, ML. Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol Genet Metab 2003, 80, 148–158. [Google Scholar]
- Check, E. Gene therapy put on hold as third child develops cancer. Nature 2005, 433, 561. [Google Scholar]
- Luo, D; Saltzman, WM. Synthetic DNA delivery systems. Nat Biotechnol 2000, 18, 33–37. [Google Scholar]
- Frankel, AD; Pabo, CO. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar]
- Green, M; Loewenstein, PM. Autonomous functional domains of chemically synthesized human immunodeficiency virus tat trans-activator protein. Cell 1988, 55, 1179–1188. [Google Scholar]
- Joliot, AH; Triller, A; Volovitch, M; Pernelle, C; Prochiantz, A. alpha-2,8-Polysialic acid is the neuronal surface receptor of antennapedia homeobox peptide. New Biol 1991, 3, 1121–1134. [Google Scholar]
- Fawell, S; Seery, J; Daikh, Y; Moore, C; Chen, LL; Pepinsky, B; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA 1994, 91, 664–668. [Google Scholar]
- Allinquant, B; Hantraye, P; Mailleux, P; Moya, K; Bouillot, C; Prochiantz, A. Downregulation of amyloid precursor protein inhibits neurite outgrowth in vitro. J Cell Biol 1995, 128, 919–927. [Google Scholar]
- Schwarze, SR; Ho, A; Vocero-Akbani, A; Dowdy, SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar]
- Langel, Ü (Ed.) 2002; Cell-Penetrating Peptides: Processes and Applications; CRC Press: Boca Raton.
- Zatsepin, TS; Turner, JJ; Oretskaya, TS; Gait, MJ. Conjugates of oligonucleotides and analogues with cell penetrating peptides as gene silencing agents. Curr Pharm Des 2005, 11, 3639–3654. [Google Scholar]
- Derossi, D; Calvet, S; Trembleau, A; Brunissen, A; Chassaing, G; Prochiantz, A. Cell internalization of the third helix of the Antennapedia homeodomain is receptor-independent. J Biol Chem 1996, 271, 18188–18193. [Google Scholar]
- Morris, MC; Vidal, P; Chaloin, L; Heitz, F; Divita, G. A new peptide vector for efficient delivery of oligonucleotides into mammalian cells. Nucleic Acids Res 1997, 25, 2730–2736. [Google Scholar]
- Vives, E; Brodin, P; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem 1997, 272, 16010–16017. [Google Scholar]
- Schwarze, SR; Dowdy, SF. In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol Sci 2000, 21, 45–48. [Google Scholar]
- Futaki, S; Suzuki, T; Ohashi, W; Yagami, T; Tanaka, S; Ueda, K; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J Biol Chem 2001, 276, 5836–5840. [Google Scholar]
- Pichon, C; Monsigny, M; Roche, AC. Intracellular localization of oligonucleotides: influence of fixative protocols. Antisense Nucleic Acid Drug Dev 1999, 9, 89–93. [Google Scholar]
- Lundberg, M; Johansson, M. Is VP22 nuclear homing an artifact? Nat Biotechnol 2001, 19, 713–714. [Google Scholar]
- Lundberg, M; Johansson, M. Positively charged DNA-binding proteins cause apparent cell membrane translocation. Biochem Biophys Res Commun 2002, 291, 367–371. [Google Scholar]
- Richard, JP; Melikov, K; Vives, E; Ramos, C; Verbeure, B; Gait, MJ; Chernomordik, LV; Lebleu, B. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem 2003, 278, 585–590. [Google Scholar]
- Drin, G; Cottin, S; Blanc, E; Rees, AR; Temsamani, J. Studies on the internalization mechanism of cationic cell-penetrating peptides. J Biol Chem 2003, 278, 31192–31201. [Google Scholar]
- Rusnati, M; Coltrini, D; Oreste, P; Zoppetti, G; Albini, A; Noonan, D; d'Adda, dF; Giacca, M; Presta, M. Interaction of HIV-1 Tat protein with heparin. Role of the backbone structure, sulfation, and size. J Biol Chem 1997, 272, 11313–11320. [Google Scholar]
- Rusnati, M; Tulipano, G; Spillmann, D; Tanghetti, E; Oreste, P; Zoppetti, G; Giacca, M; Presta, M. Multiple interactions of HIV-I Tat protein with size-defined heparin oligosaccharides. J Biol Chem 1999, 274, 28198–28205. [Google Scholar]
- Tyagi, M; Rusnati, M; Presta, M; Giacca, M. Internalization of HIV-1 tat requires cell surface heparan sulfate proteoglycans. J Biol Chem 2001, 276, 3254–3261. [Google Scholar]
- Console, S; Marty, C; Garcia-Echeverria, C; Schwendener, R; Ballmer-Hofer, K. Antennapedia and HIV transactivator of transcription (TAT) “protein transduction domains” promote endocytosis of high molecular weight cargo upon binding to cell surface glycosaminoglycans. J Biol Chem 2003, 278, 35109–35114. [Google Scholar]
- Conner, SD; Schmid, SL. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar]
- Le Roy, C; Wrana, JL. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat Rev Mol Cell Biol 2005, 6, 112–126. [Google Scholar]
- Brodsky, FM; Chen, CY; Knuehl, C; Towler, MC; Wakeham, DE. Biological basket weaving: formation and function of clathrin-coated vesicles. Annu Rev Cell Dev Biol 2001, 17, 517–568. [Google Scholar]
- Sieczkarski, SB; Whittaker, GR. Dissecting virus entry via endocytosis. J Gen Virol 2002, 83, 1535–1545. [Google Scholar]
- Parton, RG; Simons, K. The multiple faces of caveolae. Nat Rev Mol Cell Biol 2007, 8, 185–194. [Google Scholar]
- Hill, MM; Bastiani, M; Luetterforst, R; Kirkham, M; Kirkham, A; Nixon, SJ; Walser, P; Abankwa, D; Oorschot, VM; Martin, S; Hancock, JF; Parton, RG. PTRF-Cavin, a conserved cytoplasmic protein required for caveola formation and function. Cell 2008, 132, 113–124. [Google Scholar]
- Sabharanjak, S; Sharma, P; Parton, RG; Mayor, S. GPI-anchored proteins are delivered to recycling endosomes via a distinct cdc42-regulated, clathrin-independent pinocytic pathway. Dev Cell 2002, 2, 411–423. [Google Scholar]
- Kirkham, M; Parton, RG. Clathrin-independent endocytosis: New insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta 2005, 1745, 273–286. [Google Scholar]
- Pelkmans, L; Fava, E; Grabner, H; Hannus, M; Habermann, B; Krausz, E; Zerial, M. Genome-wide analysis of human kinases in clathrin- and caveolae/raft-mediated endocytosis. Nature 2005, 436, 78–86. [Google Scholar]
- Richard, JP; Melikov, K; Brooks, H; Prevot, P; Lebleu, B; Chernomordik, LV. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem 2005, 280, 15300–15306. [Google Scholar]
- Fittipaldi, A; Ferrari, A; Zoppe, M; Arcangeli, C; Pellegrini, V; Beltram, F; Giacca, M. Cell membrane lipid rafts mediate caveolar endocytosis of HIV-1 Tat fusion proteins. J Biol Chem 2003, 278, 34141–34149. [Google Scholar]
- Wadia, JS; Stan, RV; Dowdy, SF. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat Med 2004, 10, 310–315. [Google Scholar]
- Terrone, D; Sang, SL; Roudaia, L; Silvius, JR. Penetratin and related cell-penetrating cationic peptides can translocate across lipid bilayers in the presence of a transbilayer potential. Biochemistry 2003, 42, 13787–13799. [Google Scholar]
- Deshayes, S; Heitz, A; Morris, MC; Charnet, P; Divita, G; Heitz, F. Insight into the mechanism of internalization of the cell-penetrating carrier peptide Pep-1 through conformational analysis. Biochemistry 2004, 43, 1449–1457. [Google Scholar]
- Deshayes, S; Gerbal-Chaloin, S; Morris, MC; Aldrian-Herrada, G; Charnet, P; Divita, G; Heitz, F. On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids. Biochim Biophys Acta 2004, 1667, 141–147. [Google Scholar]
- Henriques, ST; Castanho, MA. Consequences of nonlytic membrane perturbation to the translocation of the cell penetrating peptide pep-1 in lipidic vesicles. Biochemistry 2004, 43, 9716–9724. [Google Scholar]
- Thoren, PE; Persson, D; Esbjorner, EK; Goksor, M; Lincoln, P; Norden, B. Membrane binding and translocation of cell-penetrating peptides. Biochemistry 2004, 43, 3471–3489. [Google Scholar]
- Simons, K; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar]
- Rothbard, J; Jessop, TC; Lewis, RS; Murray, BA; Wender, PA. Role of membrane potential and hydrogen bonding in the mechanism of translocation of guanidinium-rich peptides into cells. J Am Chem Soc 2004, 126, 9506–9507. [Google Scholar]
- Maiolo, JR; Ferrer, M; Ottinger, EA. Effects of cargo molecules on the cellular uptake of arginine-rich cell-penetrating peptides. Biochim Biophys Acta 2005, 1712, 161–172. [Google Scholar]
- Mano, M; Teodosio, C; Paiva, A; Simoes, S; Pedroso de Lima, MC. On the mechanisms of the internalization of S4(13)-PV cell-penetrating peptide. Biochem J 2005, 390, 603–612. [Google Scholar]
- De Coupade, C; Fittipaldi, A; Chagnas, V; Michel, M; Carlier, S; Tasciotti, E; Darmon, A; Ravel, D; Kearsey, J; Giacca, M; Cailler, F. Novel human-derived cell-penetrating peptides for specific subcellular delivery of therapeutic biomolecules. Biochem J 2005, 390, 407–418. [Google Scholar]
- El-Andaloussi, S. A novel cell-penetrating peptide, M918, for efficient delivery of proteins and peptide nucleic acids. Mol Ther 2007, 15, 1820–1826. [Google Scholar]
- Trehin, R; Merkle, HP. Chances and pitfalls of cell penetrating peptides for cellular drug delivery. Eur J Pharm Biopharm 2004, 58, 209–223. [Google Scholar]
- Fischer, R; Fotin-Mleczek, M; Hufnagel, H; Brock, R. Break on through to the other side-biophysics and cell biology shed light on cell-penetrating peptides. Chembiochem 2005, 6, 2126–2142. [Google Scholar]
- Melikov, K; Chernomordik, LV. Arginine-rich cell penetrating peptides: from endosomal uptake to nuclear delivery. Cell Mol Life Sci 2005, 62, 2739–2749. [Google Scholar]
- Vives, E. Present and future of cell-penetrating peptide mediated delivery systems: “Is the Trojan horse too wild to go only to Troy?”. J Control Release 2005, 109, 77–85. [Google Scholar]
- Langel, Ü (Ed.) 2006; Handbook of Cell-Penetrating Peptides; CRC Press: Boca Raton.
- Lochmann, D; Jauk, E; Zimmer, A. Drug delivery of oligonucleotides by peptides. Eur J Pharm Biopharm 2004, 58, 237–251. [Google Scholar]
- Dietz, GP; Bahr, M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci 2004, 27, 85–131. [Google Scholar]
- Järver, P; Langel, K; El-Andaloussi, S; Langel, Ü. Applications of cell-penetrating peptides in regulation of gene expression. Biochem Soc Trans 2007, 35, 770–774. [Google Scholar]
- Derossi, D; Joliot, AH; Chassaing, G; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J Biol Chem 1994, 269, 10444–10450. [Google Scholar]
- Pooga, M; Hällbrink, M; Zorko, M; Langel, Ü. Cell penetration by transportan. FASEB J 1998, 12, 67–77. [Google Scholar]
- Soomets, U; Lindgren, M; Gallet, X; Hällbrink, M; Elmquist, A; Balaspiri, L; Zorko, M; Pooga, M; Brasseur, R; Langel, Ü. Deletion analogues of transportan. Biochim Biophys Acta 2000, 1467, 165–176. [Google Scholar]
- Mitchell, DJ; Kim, DT; Steinman, L; Fathman, CG; Rothbard, JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res 2000, 56, 318–325. [Google Scholar]
- Oehlke, J; Scheller, A; Wiesner, B; Krause, E; Beyermann, M; Klauschenz, E; Melzig, M; Bienert, M. Cellular uptake of an alpha-helical amphipathic model peptide with the potential to deliver polar compounds into the cell interior non-endocytically. Biochim Biophys Acta 1998, 1414, 127–139. [Google Scholar]
- Deshayes, S; Plenat, T; Aldrian-Herrada, G; Divita, G; Le Grimellec, C; Heitz, F. Primary amphipathic cell-penetrating peptides: structural requirements and interactions with model membranes. Biochemistry 2004, 43, 7698–7706. [Google Scholar]
- Krauss, U; Müller, M; Stahl, M; Beck-Sickinger, AG. In vitro gene delivery by a novel human calcitonin (hCT)-derived carrier peptide. Bioorg Med Chem Lett 2004, 14, 51–54. [Google Scholar]
- Fernandez-Carneado, J; Kogan, MJ; Castel, S; Giralt, E. Potential peptide carriers: amphipathic proline-rich peptides derived from the N-terminal domain of gamma-zein. Angew Chem Int Ed Engl 2004, 43, 1811–1814. [Google Scholar]
- Hariton-Gazal, E; Feder, R; Mor, A; Graessmann, A; Brack-Werner, R; Jans, D; Gilon, C; Loyter, A. Targeting of nonkaryophilic cell-permeable peptides into the nuclei of intact cells by covalently attached nuclear localization signals. Biochemistry 2002, 41, 9208–9214. [Google Scholar]
- Lundberg, P; Magzoub, M; Lindberg, M; Hällbrink, M; Jarvet, J; Eriksson, LE; Langel, Ü; Gräslund, A. Cell membrane translocation of the N-terminal (1–28) part of the prion protein. Biochem Biophys Res Commun 2002, 299, 85–90. [Google Scholar]
- Magzoub, M; Oglecka, K; Pramanik, A; Goran Eriksson, LE; Gräslund, A. Membrane perturbation effects of peptides derived from the N-termini of unprocessed prion proteins. Biochim Biophys Acta 2005, 1716, 126–136. [Google Scholar]
- Biverstahl, H; Andersson, A; Gräslund, A; Maler, L. NMR solution structure and membrane interaction of the N-terminal sequence (1–30) of the bovine prion protein. Biochemistry 2004, 43, 14940–14947. [Google Scholar]
- Gomez, JA; Gama, V; Yoshida, T; Sun, W; Hayes, P; Leskov, K; Boothman, D; Matsuyama, S. Bax-inhibiting peptides derived from Ku70 and cell-penetrating pentapeptides. Biochem Soc Trans 2007, 35, 797–801. [Google Scholar]
- Lundberg, P; El-Andaloussi, S; Sutlu, T; Johansson, H; Langel, Ü. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J 2007, 21, 2664–2671. [Google Scholar]
- Brooks, H; Lebleu, B; Vives, E. Tat peptide-mediated cellular delivery: back to basics. Adv Drug Deliv Rev 2005, 57, 559–577. [Google Scholar]
- Wadia, JS; Dowdy, SF. Transmembrane delivery of protein and peptide drugs by TAT-mediated transduction in the treatment of cancer. Adv Drug Deliv Rev 2005, 57, 579–596. [Google Scholar]
- Gupta, B; Torchilin, VP. Transactivating transcriptional activator-mediated drug delivery. Expert Opin Drug Deliv 2006, 3, 177–190. [Google Scholar]
- Torchilin, VP. Tat peptide-mediated intracellular delivery of pharmaceutical nanocarriers. Adv Drug Deliv Rev 2008, 60, 548–558. [Google Scholar]
- Wender, PA; Mitchell, DJ; Pattabiraman, K; Pelkey, ET; Steinman, L; Rothbard, JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci USA 2000, 97, 13003–13008. [Google Scholar]
- Chen, YN; Sharma, SK; Ramsey, TM; Jiang, L; Martin, MS; Baker, K; Adams, PD; Bair, KW; Kaelin, WG, Jr. Selective killing of transformed cells by cyclin/cyclin-dependent kinase 2 antagonists. Proc Natl Acad Sci USA 1999, 96, 4325–4329. [Google Scholar]
- Arnt, CR; Chiorean, MV; Heldebrant, MP; Gores, GJ; Kaufmann, SH. Synthetic Smac/DIABLO peptides enhance the effects of chemotherapeutic agents by binding XIAP and cIAP1 in situ. J Biol Chem 2002, 277, 44236–44243. [Google Scholar]
- Asada, S; Choi, Y; Yamada, M; Wang, SC; Hung, MC; Qin, J; Uesugi, M. External control of Her2 expression and cancer cell growth by targeting a Ras-linked coactivator. Proc Natl Acad Sci USA 2002, 99, 12747–12752. [Google Scholar]
- Koppelhus, U; Awasthi, SK; Zachar, V; Holst, HU; Ebbesen, P; Nielsen, PE. Cell-dependent differential cellular uptake of PNA, peptides, and PNA-peptide conjugates. Antisense Nucleic Acid Drug Dev 2002, 12, 51–63. [Google Scholar]
- Astriab-Fisher, A; Sergueev, DS; Fisher, M; Shaw, BR; Juliano, RL. Antisense inhibition of P-glycoprotein expression using peptide-oligonucleotide conjugates. Biochem Pharmacol 2000, 60, 83–90. [Google Scholar]
- Astriab-Fisher, A; Sergueev, D; Fisher, M; Shaw, BR; Juliano, RL. Conjugates of antisense oligonucleotides with the Tat and antennapedia cell-penetrating peptides: effects on cellular uptake, binding to target sequences, and biologic actions. Pharm Res 2002, 19, 744–754. [Google Scholar]
- Moulton, HM; Moulton, JD. Peptide-assisted delivery of steric-blocking antisense oligomers. Curr Opin Mol Ther 2003, 5, 123–132. [Google Scholar]
- Moulton, HM; Hase, MC; Smith, KM; Iversen, PL. HIV Tat peptide enhances cellular delivery of antisense morpholino oligomers. Antisense Nucleic Acid Drug Dev 2003, 13, 31–43. [Google Scholar]
- Chiu, YL; Ali, A; Chu, CY; Cao, H; Rana, TM. Visualizing a correlation between siRNA localization, cellular uptake, and RNAi in living cells. Chem Biol 2004, 11, 1165–1175. [Google Scholar]
- Sandgren, S; Cheng, F; Belting, M. Nuclear targeting of macromolecular polyanions by an HIV-Tat derived peptide. Role for cell-surface proteoglycans. J Biol Chem 2002, 277, 38877–38883. [Google Scholar]
- Ignatovich, IA; Dizhe, EB; Pavlotskaya, AV; Akifiev, BN; Burov, SV; Orlov, SV; Perevozchikov, AP. Complexes of plasmid DNA with basic domain 47–57 of the HIV-1 Tat protein are transferred to mammalian cells by endocytosis-mediated pathways. J Biol Chem 2003, 278, 42625–42636. [Google Scholar]
- Rudolph, C; Plank, C; Lausier, J; Schillinger, U; Müller, RH; Rosenecker, J. Oligomers of the arginine-rich motif of the HIV-1 TAT protein are capable of transferring plasmid DNA into cells. J Biol Chem 2003, 278, 11411–11418. [Google Scholar]
- Torchilin, VP; Rammohan, R; Weissig, V; Levchenko, TS. TAT peptide on the surface of liposomes affords their efficient intracellular delivery even at low temperature and in the presence of metabolic inhibitors. Proc Natl Acad Sci USA 2001, 98, 8786–8791. [Google Scholar]
- Tseng, YL; Liu, JJ; Hong, RL. Translocation of liposomes into cancer cells by cell-penetrating peptides penetratin and tat: a kinetic and efficacy study. Mol Pharmacol 2002, 62, 864–872. [Google Scholar]
- Torchilin, VP; Levchenko, TS; Rammohan, R; Volodina, N. Papahadjopoulos-Sternberg, B.; D'Souza, G. G. Cell transfection in vitro and in vivo with nontoxic TAT peptide-liposome-DNA complexes. Proc Natl Acad Sci USA 2003, 100, 1972–1977. [Google Scholar]
- Gratton, JP; Yu, J; Griffith, JW; Babbitt, RW; Scotland, RS; Hickey, R; Giordano, FJ; Sessa, WC. Cell-permeable peptides improve cellular uptake and therapeutic gene delivery of replication-deficient viruses in cells and in vivo. Nat Med 2003, 9, 357–362. [Google Scholar]
- Prochiantz, A. Getting hydrophilic compounds into cells: lessons from homeopeptides. Curr Opin Neurobiol 1996, 6, 629–634. [Google Scholar]
- Drin, G; Mazel, M; Clair, P; Mathieu, D; Kaczorek, M; Temsamani, J. Physico-chemical requirements for cellular uptake of pAntp peptide. Role of lipid-binding affinity. Eur J Biochem 2001, 268, 1304–1314. [Google Scholar]
- Bárány-Wallje, E; Keller, S; Serowy, S; Geibel, S; Pohl, P; Bienert, M; Dathe, M. A Critical Reassessment of Penetratin Translocation Across Lipid Membranes. Biophys J 2005. [Google Scholar]
- Derossi, D; Williams, EJ; Green, PJ; Dunican, DJ; Doherty, P. Stimulation of mitogenesis by a cell-permeable PI 3-kinase binding peptide. Biochem Biophys Res Commun 1998, 251, 148–152. [Google Scholar]
- Joliot, A; Prochiantz, A. Transduction peptides: from technology to physiology. Nat Cell Biol 2004, 6, 189–196. [Google Scholar]
- Joliot, A; Prochiantz, A. Homeoproteins as natural Penetratin cargoes with signaling properties. Adv Drug Deliv Rev 2008, 60, 608–613. [Google Scholar]
- Muratovska, A; Eccles, MR. Conjugate for efficient delivery of short interfering RNA (siRNA) into mammalian cells. FEBS Lett 2004, 558, 63–68. [Google Scholar]
- Bendifallah, N; Rasmussen, FW; Zachar, V; Ebbesen, P; Nielsen, PE; Koppelhus, U. Evaluation of cell-penetrating peptides (CPPs) as vehicles for intracellular delivery of antisense peptide nucleic acid (PNA). Bioconjug Chem 2006, 17, 750–758. [Google Scholar]
- Moschos, SA; Jones, SW; Perry, MM; Williams, AE; Erjefalt, JS; Turner, JJ; Barnes, PJ; Sproat, BS; Gait, MJ; Lindsay, MA. Lung delivery studies using siRNA conjugated to TAT(48–60) and penetratin reveal peptide induced reduction in gene expression and induction of innate immunity. Bioconjug Chem 2007, 18, 1450–1459. [Google Scholar]
- Chaubey, B; Tripathi, S; Pandey, VN. Single Acute-Dose and Repeat-Doses Toxicity of anti-HIV-1 PNA(TAR)-Penetratin Conjugate after Intraperitoneal Administration to Mice. Oligonucleotides 2008, 18, 9–20. [Google Scholar]
- Fabani, MM; Gait, MJ. miR-122 targeting with LNA/2′-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA 2008, 14, 336–346. [Google Scholar]
- Abes, S; Turner, JJ; Ivanova, GD; Owen, D; Williams, D; Arzumanov, A; Clair, P; Gait, MJ; Lebleu, B. Efficient splicing correction by PNA conjugation to an R6-Penetratin delivery peptide. Nucleic Acids Res 2007, 35, 4495–4502. [Google Scholar]
- Pooga, M; Kut, C; Kihlmark, M; Hällbrink, M; Fernaeus, S; Raid, R; Land, T; Hallberg, E; Bartfai, T; Langel, Ü. Cellular translocation of proteins by transportan. FASEB J 2001, 15, 1451–1453. [Google Scholar]
- Kilk, K; El-Andaloussi, S; Järver, P; Meikas, A; Valkna, A; Bartfai, T; Kogerman, P; Metsis, M; Langel, Ü. Evaluation of transportan 10 in PEI mediated plasmid delivery assay. J Control Release 2005, 103, 511–523. [Google Scholar]
- Pooga, M; Soomets, U; Hällbrink, M; Valkna, A; Saar, K; Rezaei, K; Kahl, U; Hao, JX; Xu, XJ; Wiesenfeld-Hallin, Z; Hokfelt, T; Bartfai, T; Langel, Ü. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat Biotechnol 1998, 16, 857–861. [Google Scholar]
- Fisher, L; Soomets, U; Cortes, TV; Chilton, L; Jiang, Y; Langel, Ü; Iverfeldt, K. Cellular delivery of a double-stranded oligonucleotide NFkappaB decoy by hybridization to complementary PNA linked to a cell-penetrating peptide. Gene Ther 2004, 11, 1264–1272. [Google Scholar]
- Turner, JJ; Jones, S; Fabani, MM; Ivanova, G; Arzumanov, AA; Gait, MJ. RNA targeting with peptide conjugates of oligonucleotides, siRNA and PNA. Blood Cells Mol Dis 2007, 38, 1–7. [Google Scholar]
- El-Andaloussi, S; Johansson, HJ; Lundberg, P; Langel, Ü. Induction of splice correction by cell-penetrating peptide nucleic acids. J Gene Med 2006, 8, 1262–1273. [Google Scholar]
- Padari, K; Säälik, P; Hansen, M; Koppel, K; Raid, R; Langel, Ü; Pooga, M. Cell transduction pathways of transportans. Bioconjug Chem 2005, 16, 1399–1410. [Google Scholar]
- Futaki, S. Oligoarginine vectors for intracellular delivery: design and cellular-uptake mechanisms. Biopolymers 2006, 84, 241–249. [Google Scholar]
- Futaki, S; Nakase, I; Suzuki, T; Youjun, Z; Sugiura, Y. Translocation of branched-chain arginine peptides through cell membranes: flexibility in the spatial disposition of positive charges in membrane-permeable peptides. Biochemistry 2002, 41, 7925–7930. [Google Scholar]
- Futaki, S; Niwa, M; Nakase, I; Tadokoro, A; Zhang, Y; Nagaoka, M; Wakako, N; Sugiura, Y. Arginine carrier peptide bearing Ni(II) chelator to promote cellular uptake of histidine-tagged proteins. Bioconjug Chem 2004, 15, 475–481. [Google Scholar]
- Kish, PE; Tsume, Y; Kijek, P; Lanigan, TM; Hilfinger, JM; Roessler, BJ. Bile acid-oligopeptide conjugates interact with DNA and facilitate transfection. Mol Pharm 2007, 4, 95–103. [Google Scholar]
- Kim, WJ; Christensen, LV; Jo, S; Yockman, JW; Jeong, JH; Kim, YH; Kim, SW. Cholesteryl oligoarginine delivering vascular endothelial growth factor siRNA effectively inhibits tumor growth in colon adenocarcinoma. Mol Ther 2006, 14, 343–350. [Google Scholar]
- Nakase, I; Niwa, M; Takeuchi, T; Sonomura, K; Kawabata, N; Koike, Y; Takehashi, M; Tanaka, S; Ueda, K; Simpson, JC; Jones, AT; Sugiura, Y; Futaki, S. Cellular Uptake of Arginine-Rich Peptides: Roles for Macropinocytosis and Actin Rearrangement. Mol Ther 2004, 10, 1011–1022. [Google Scholar]
- Nakase, I; Tadokoro, A; Kawabata, N; Takeuchi, T; Katoh, H; Hiramoto, K; Negishi, M; Nomizu, M; Sugiura, Y; Futaki, S. Interaction of arginine-rich peptides with membrane-associated proteoglycans is crucial for induction of actin organization and macropinocytosis. Biochemistry 2007, 46, 492–501. [Google Scholar]
- Fischer, R; Kohler, K; Fotin-Mleczek, M; Brock, R. A stepwise dissection of the intracellular fate of cationic cell-penetrating peptides. J Biol Chem 2004, 279, 12625–12635. [Google Scholar]
- Steiner, V; Schar, M; Bornsen, KO; Mutter, M. Retention behaviour of a template-assembled synthetic protein and its amphiphilic building blocks on reversed-phase columns. J Chromatogr 1991, 586, 43–50. [Google Scholar]
- Oehlke, J; Birth, P; Klauschenz, E; Wiesner, B; Beyermann, M; Oksche, A; Bienert, M. Cellular uptake of antisense oligonucleotides after complexing or conjugation with cell-penetrating model peptides. Eur J Biochem 2002, 269, 4025–4032. [Google Scholar]
- Oehlke, J; Wallukat, G; Wolf, Y; Ehrlich, A; Wiesner, B; Berger, H; Bienert, M. Enhancement of intracellular concentration and biological activity of PNA after conjugation with a cell-penetrating synthetic model peptide. Eur J Biochem 2004, 271, 3043–3049. [Google Scholar]
- Wolf, Y; Pritz, S; Abes, S; Bienert, M; Lebleu, B; Oehlke, J. Structural requirements for cellular uptake and antisense activity of peptide nucleic acids conjugated with various peptides. Biochemistry 2006, 45, 14944–14954. [Google Scholar]
- Shiraishi, T; Nielsen, PE. Photochemically enhanced cellular delivery of cell penetrating peptide-PNA conjugates. FEBS Lett 2006, 580, 1451–1456. [Google Scholar]
- Morris, MC; Chaloin, L; Mery, J; Heitz, F; Divita, G. A novel potent strategy for gene delivery using a single peptide vector as a carrier. Nucleic Acids Res 1999, 27, 3510–3517. [Google Scholar]
- Simeoni, F; Morris, MC; Heitz, F; Divita, G. Insight into the mechanism of the peptide-based gene delivery system MPG: implications for delivery of siRNA into mammalian cells. Nucleic Acids Res 2003, 31, 2717–2724. [Google Scholar]
- Morris, MC; Depollier, J; Mery, J; Heitz, F; Divita, G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol 2001, 19, 1173–1176. [Google Scholar]
- Morris, MC; Chaloin, L; Choob, M; Archdeacon, J; Heitz, F; Divita, G. Combination of a new generation of PNAs with a peptide-based carrier enables efficient targeting of cell cycle progression. Gene Ther 2004, 11, 757–764. [Google Scholar]
- Morris, MC; Gros, E; Aldrian-Herrada, G; Choob, M; Archdeacon, J; Heitz, F; Divita, G. A non-covalent peptide-based carrier for in vivo delivery of DNA mimics. Nucleic Acids Res 2007, 35, e49. [Google Scholar]
- Morris, MC; Deshayes, S; Heitz, F; Divita, G. Cell-penetrating peptides: from molecular mechanisms to therapeutics. Biol Cell 2008, 100, 201–217. [Google Scholar]
- Deshayes, S; Morris, MC; Divita, G; Heitz, F. Cell-penetrating peptides: tools for intracellular delivery of therapeutics. Cell Mol Life Sci 2005, 62, 1839–1849. [Google Scholar]
- Gerbal-Chaloin, S; Gondeau, C; Aldrian-Herrada, G; Heitz, F; Gauthier-Rouviere, C; Divita, G. First step of the cell-penetrating peptide mechanism involves Rac1 GTPase-dependent actin-network remodelling. Biol Cell 2007, 99, 223–238. [Google Scholar]
- Zaidi, M; Inzerillo, AM; Moonga, BS; Bevis, PJ; Huang, CL. Forty years of calcitonin--where are we now? A tribute to the work of Iain Macintyre, FRS. Bone 2002, 30, 655–663. [Google Scholar]
- Pontiroli, AE; Alberetto, M; Pozza, G. Intranasal calcitonin and plasma calcium concentrations in normal subjects. Br Med J (Clin Res Ed) 1985, 290, 1390–1391. [Google Scholar]
- Machova, Z; Muhle, C; Krauss, U; Trehin, R; Koch, A; Merkle, HP; Beck-Sickinger, AG. Cellular internalization of enhanced green fluorescent protein ligated to a human calcitonin-based carrier peptide. Chembiochem 2002, 3, 672–677. [Google Scholar]
- Foerg, C; Ziegler, U; Fernandez-Carneado, J; Giralt, E; Rennert, R; Beck-Sickinger, AG; Merkle, HP. Decoding the Entry of Two Novel Cell-Penetrating Peptides in HeLa Cells: Lipid Raft-Mediated Endocytosis and Endosomal Escape. Biochemistry 2005, 44, 72–81. [Google Scholar]
- Rennert, R; Neundorf, I; Jahnke, HG; Suchowerskyj, P; Dournaud, P; Robitzki, A; Beck-Sickinger, AG. Generation of Carrier Peptides for the Delivery of Nucleic Acid Drugs in Primary Cells. ChemMedChem 2008. [Google Scholar]
- Rennert, R; Wespe, C; Beck-Sickinger, AG; Neundorf, I. Developing novel hCT derived cell-penetrating peptides with improved metabolic stability. Biochim Biophys Acta 2006, 1758, 347–354. [Google Scholar]
- Rennert, R; Neundorf, I; Beck-Sickinger, AG. Calcitonin-derived peptide carriers: Mechanisms and application. Adv Drug Deliv Rev 2008, 60, 485–498. [Google Scholar]
- Kogan, MJ; Dalcol, I; Gorostiza, P; Lopez-Iglesias, C; Pons, R; Pons, M; Sanz, F; Giralt, E. Supramolecular properties of the proline-rich gamma-Zein N-terminal domain. Biophys J 2002, 83, 1194–1204. [Google Scholar]
- Kogan, MJ; Lopez, O; Cocera, M; Lopez-Iglesias, C; De La, MA; Giralt, E. Exploring the interaction of the surfactant N-terminal domain of gamma-Zein with soybean phosphatidylcholine liposomes. Biopolymers 2004, 73, 258–268. [Google Scholar]
- Fernandez-Carneado, J; Kogan, MJ; Van Mau, N; Pujals, S; Lopez-Iglesias, C; Heitz, F; Giralt, E. Fatty acyl moieties: improving Pro-rich peptide uptake inside HeLa cells. J Pept Res 2005, 65, 580–590. [Google Scholar]
- Pujals, S; Fernandez-Carneado, J; Kogan, MJ; Martinez, J; Cavelier, F; Giralt, E. Replacement of a proline with silaproline causes a 20-fold increase in the cellular uptake of a Pro-rich peptide. J Am Chem Soc 2006, 128, 8479–8483. [Google Scholar]
- Pujals, S; Fernandez-Carneado, J; Ludevid, MD; Giralt, E. D-SAP: A New, Noncytotoxic, and Fully Protease Resistant Cell-Penetrating Peptide. ChemMedChem 2007. [Google Scholar]
- Pujals, S; Sabido, E; Tarrago, T; Giralt, E. all-D proline-rich cell-penetrating peptides: a preliminary in vivo internalization study. Biochem Soc Trans 2007, 35, 794–796. [Google Scholar]
- Mor, A; Nguyen, VH; Delfour, A; Migliore-Samour, D; Nicolas, P. Isolation, amino acid sequence, and synthesis of dermaseptin, a novel antimicrobial peptide of amphibian skin. Biochemistry 1991, 30, 8824–8830. [Google Scholar]
- Mor, A; Amiche, M; Nicolas, P. Structure, synthesis, and activity of dermaseptin b, a novel vertebrate defensive peptide from frog skin: relationship with adenoregulin. Biochemistry 1994, 33, 6642–6650. [Google Scholar]
- Mor, A; Nicolas, P. Isolation and structure of novel defensive peptides from frog skin. Eur J Biochem 1994, 219, 145–154. [Google Scholar]
- Mor, A; Nicolas, P. The NH2-terminal alpha-helical domain 1–18 of dermaseptin is responsible for antimicrobial activity. J Biol Chem 1994, 269, 1934–1939. [Google Scholar]
- Pouny, Y; Rapaport, D; Mor, A; Nicolas, P; Shai, Y. Interaction of antimicrobial dermaseptin and its fluorescently labeled analogues with phospholipid membranes. Biochemistry 1992, 31, 12416–12423. [Google Scholar]
- Strahilevitz, J; Mor, A; Nicolas, P; Shai, Y. Spectrum of antimicrobial activity and assembly of dermaseptin-b and its precursor form in phospholipid membranes. Biochemistry 1994, 33, 10951–10960. [Google Scholar]
- Ghosh, JK; Shaool, D; Guillaud, P; Ciceron, L; Mazier, D; Kustanovich, I; Shai, Y; Mor, A. Selective cytotoxicity of dermaseptin S3 toward intraerythrocytic Plasmodium falciparum and the underlying molecular basis. J Biol Chem 1997, 272, 31609–31616. [Google Scholar]
- Mano, M; Henriques, A; Paiva, A; Prieto, M; Gavilanes, F; Simoes, S; Pedroso de Lima, MC. Cellular uptake of S413-PV peptide occurs upon conformational changes induced by peptide-membrane interactions. Biochim Biophys Acta 2006, 1758, 336–346. [Google Scholar]
- Mano, M; Henriques, A; Paiva, A; Prieto, M; Gavilanes, F; Simoes, S; de Lima, MC. Interaction of S413-PV cell penetrating peptide with model membranes: relevance to peptide translocation across biological membranes. J Pept Sci 2007, 13, 301–313. [Google Scholar]
- Magzoub, M; Pramanik, A; Gräslund, A. Modeling the endosomal escape of cell-penetrating peptides: transmembrane pH gradient driven translocation across phospholipid bilayers. Biochemistry 2005, 44, 14890–14897. [Google Scholar]
- Magzoub, M; Sandgren, S; Lundberg, P; Oglecka, K; Lilja, J; Wittrup, A; Goran Eriksson, LE; Langel, Ü; Belting, M; Gräslund, A. N-terminal peptides from unprocessed prion proteins enter cells by macropinocytosis. Biochem Biophys Res Commun 2006, 348, 379–385. [Google Scholar]
- Yoshida, T; Tomioka, I; Nagahara, T; Holyst, T; Sawada, M; Hayes, P; Gama, V; Okuno, M; Chen, Y; Abe, Y; Kanouchi, T; Sasada, H; Wang, D; Yokota, T; Sato, E; Matsuyama, S. Bax-inhibiting peptide derived from mouse and rat Ku70. Biochem Biophys Res Commun 2004, 321, 961–966. [Google Scholar]
- Sawada, M; Hayes, P; Matsuyama, S. Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat Cell Biol 2003, 5, 352–357. [Google Scholar]
- Preuss, M; Tecle, M; Shah, I; Matthews, DA; Miller, AD. Comparison between the interactions of adenovirus-derived peptides with plasmid DNA and their role in gene delivery mediated by liposome-peptide-DNA virus-like nanoparticles. Org Biomol Chem 2003, 1, 2430–2438. [Google Scholar]
- Read, ML; Bremner, KH; Oupicky, D; Green, NK; Searle, PF; Seymour, LW. Vectors based on reducible polycations facilitate intracellular release of nucleic acids. J Gene Med 2003, 5, 232–245. [Google Scholar]
- Hyndman, L; Lemoine, JL; Huang, L; Porteous, DJ; Boyd, AC; Nan, X. HIV-1 Tat protein transduction domain peptide facilitates gene transfer in combination with cationic liposomes. J Control Release 2004, 99, 435–444. [Google Scholar]
- Futaki, S; Masui, Y; Nakase, I; Sugiura, Y; Nakamura, T; Kogure, K; Harashima, H. Unique features of a pH-sensitive fusogenic peptide that improves the transfection efficiency of cationic liposomes. J Gene Med 2005, 7, 1450–1458. [Google Scholar]
- Read, ML; Singh, S; Ahmed, Z; Stevenson, M; Briggs, SS; Oupicky, D; Barrett, LB; Spice, R; Kendall, M; Berry, M; Preece, JA; Logan, A; Seymour, LW. A versatile reducible polycation-based system for efficient delivery of a broad range of nucleic acids. Nucleic Acids Res 2005, 33, e86. [Google Scholar]
- Midoux, P; Monsigny, M. Efficient gene transfer by histidylated polylysine/pDNA complexes. Bioconjug Chem 1999, 10, 406–411. [Google Scholar]
- Chen, QR; Zhang, L; Stass, SA; Mixson, AJ. Branched co-polymers of histidine and lysine are efficient carriers of plasmids. Nucleic Acids Res 2001, 29, 1334–1340. [Google Scholar]
- Ritter, W; Plank, C; Lausier, J; Rudolph, C; Zink, D; Reinhardt, D; Rosenecker, J. A novel transfecting peptide comprising a tetrameric nuclear localization sequence. J Mol Med 2003, 81, 708–717. [Google Scholar]
- Leng, Q; Scaria, P; Zhu, J; Ambulos, N; Campbell, P; Mixson, AJ. Highly branched HK peptides are effective carriers of siRNA. J Gene Med 2005, 7, 977–986. [Google Scholar]
- Liu, Z; Li, M; Cui, D; Fei, J. Macro-branched cell-penetrating peptide design for gene delivery. J Control Release 2005, 102, 699–710. [Google Scholar]
- Bayele, HK; Sakthivel, T; O'Donell, M; Pasi, KJ; Wilderspin, AF; Lee, CA; Toth, I; Florence, AT. Versatile peptide dendrimers for nucleic acid delivery. J Pharm Sci 2005, 94, 446–457. [Google Scholar]
- Kang, H; Delong, R; Fisher, MH; Juliano, RL. Tat-Conjugated PAMAM Dendrimers as Delivery Agents for Antisense and siRNA Oligonucleotides. Pharm Res 2005, 22, 2099–2106. [Google Scholar]
- Bayele, HK; Ramaswamy, C; Wilderspin, AF; Srai, KS; Toth, I; Florence, AT. Protein transduction by lipidic peptide dendrimers. J Pharm Sci 2006, 95, 1227–1237. [Google Scholar]
- Kale, AA; Torchilin, VP. Design, synthesis, and characterization of pH-sensitive PEG-PE conjugates for stimuli-sensitive pharmaceutical nanocarriers: the effect of substitutes at the hydrazone linkage on the ph stability of PEG-PE conjugates. Bioconjug Chem 2007, 18, 363–370. [Google Scholar]
- Kale, AA; Torchilin, VP. “Smart” drug carriers: PEGylated TATp-modified pH-sensitive liposomes. J Liposome Res 2007, 17, 197–203. [Google Scholar]
- Khalil, IA; Kogure, K; Futaki, S; Hama, S; Akita, H; Ueno, M; Kishida, H; Kudoh, M; Mishina, Y; Kataoka, K; Yamada, M; Harashima, H. Octaarginine-modified multifunctional envelope-type nanoparticles for gene delivery. Gene Ther 2007, 14, 682–689. [Google Scholar]
- Soundara Manickam, D; Oupický, D. Multiblock reducible copolypeptides containing histidine-rich and nuclear localization sequences for gene delivery. Bioconjug Chem 2006, 17, 1395–1403. [Google Scholar]
- Rahbek, UL; Howard, KA; Oupický, D; Manickam, DS; Dong, M; Nielsen, AF; Hansen, TB; Besenbacher, F; Kjems, J. Intracellular siRNA and precursor miRNA trafficking using bioresponsive copolypeptides. J Gene Med 2008, 10, 81–93. [Google Scholar]
- Chen, QR; Zhang, L; Stass, SA; Mixson, AJ. Co-polymer of histidine and lysine markedly enhances transfection efficiency of liposomes. Gene Ther 2000, 7, 1698–1705. [Google Scholar]
- Leng, Q; Mixson, AJ. Modified branched peptides with a histidine-rich tail enhance in vitro gene transfection. Nucleic Acids Res 2005, 33, e40. [Google Scholar]
- Leng, Q; Scaria, P; Ioffe, OB; Woodle, M; Mixson, AJ. A branched histidine/lysine peptide, H2K4b, in complex with plasmids encoding antitumor proteins inhibits tumor xenografts. J Gene Med 2006, 8, 1407–1415. [Google Scholar]
- Kogure, K; Moriguchi, R; Sasaki, K; Ueno, M; Futaki, S; Harashima, H. Development of a non-viral multifunctional envelope-type nano device by a novel lipid film hydration method. J Control Release 2004, 98, 317–323. [Google Scholar]
- Khalil, IA; Kogure, K; Futaki, S; Harashima, H. High density of octaarginine stimulates macropinocytosis leading to efficient intracellular trafficking for gene expression. J Biol Chem 2006, 281, 3544–3551. [Google Scholar]
- Nakamura, Y; Kogure, K; Futaki, S; Harashima, H. Octaarginine-modified multifunctional envelope-type nano device for siRNA. J Control Release 2007, 119, 360–367. [Google Scholar]
- Nakamura, Y; Kogure, K; Yamada, Y; Futaki, S; Harashima, H. Significant and prolonged antisense effect of a multifunctional envelope-type nano device encapsulating antisense oligodeoxynucleotide. J Pharm Pharmacol 2006, 58, 431–437. [Google Scholar]
- Mudhakir, D; Akita, H; Tan, E; Harashima, H. A novel IRQ ligand-modified nano-carrier targeted to a unique pathway of caveolar endocytic pathway. J Control Release 2008, 125, 164–173. [Google Scholar]
- Enbäck, J; Laakkonen, P. Tumour-homing peptides: tools for targeting, imaging and destruction. Biochem Soc Trans 2007, 35, 780–783. [Google Scholar]
- Dash, PR; Read, ML; Barrett, LB; Wolfert, MA; Seymour, LW. Factors affecting blood clearance and in vivo distribution of polyelectrolyte complexes for gene delivery. Gene Ther 1999, 6, 643–650. [Google Scholar]
- Kobayashi, N; Kuramoto, T; Yamaoka, K; Hashida, M; Takakura, Y. Hepatic uptake and gene expression mechanisms following intravenous administration of plasmid DNA by conventional and hydrodynamics-based procedures. J Pharmacol Exp Ther 2001, 297, 853–860. [Google Scholar]
- Soundara Manickam, D; Bisht, HS; Wan, L; Mao, G; Oupický, D. Influence of TAT-peptide polymerization on properties and transfection activity of TAT/DNA polyplexes. J Control Release 2005, 102, 293–306. [Google Scholar]
- Blacklock, J; Handa, H; Soundara Manickam, D; Mao, G; Mukhopadhyay, A; Oupický, D. Disassembly of layer-by-layer films of plasmid DNA and reducible TAT polypeptide. Biomaterials 2007, 28, 117–124. [Google Scholar]
- Tung, CH; Mueller, S; Weissleder, R. Novel branching membrane translocational peptide as gene delivery vector. Bioorg Med Chem 2002, 10, 3609–3614. [Google Scholar]
- Hellgren, I; Gorman, J; Sylven, C. Factors controlling the efficiency of Tat-mediated plasmid DNA transfer. J Drug Target 2004, 12, 39–47. [Google Scholar]
- Futaki, S; Ohashi, W; Suzuki, T; Niwa, M; Tanaka, S; Ueda, K; Harashima, H; Sugiura, Y. Stearylated arginine-rich peptides: a new class of transfection systems. Bioconjug Chem 2001, 12, 1005–1011. [Google Scholar]
- Kim, TI; Baek, JU; Yoon, JK; Choi, JS; Kim, K; Park, JS. Synthesis and characterization of a novel arginine-grafted dendritic block copolymer for gene delivery and study of its cellular uptake pathway leading to transfection. Bioconjug Chem 2007, 18, 309–317. [Google Scholar]
- Crooke, ST. Progress in antisense technology. Annu Rev Med 2004, 55, 61–95. [Google Scholar]
- Chan, JH; Lim, S; Wong, WS. Antisense oligonucleotides: from design to therapeutic application. Clin Exp Pharmacol Physiol 2006, 33, 533–540. [Google Scholar]
- Troy, CM; Derossi, D; Prochiantz, A; Greene, LA; Shelanski, ML. Downregulation of Cu/Zn superoxide dismutase leads to cell death via the nitric oxide-peroxynitrite pathway. J Neurosci 1996, 16, 253–261. [Google Scholar]
- Braun, K; Peschke, P; Pipkorn, R; Lampel, S; Wachsmuth, M; Waldeck, W; Friedrich, E; Debus, J. A biological transporter for the delivery of peptide nucleic acids (PNAs) to the nuclear compartment of living cells. J Mol Biol 2002, 318, 237–243. [Google Scholar]
- Kaushik, N; Basu, A; Palumbo, P; Myers, RL; Pandey, VN. Anti-TAR polyamide nucleotide analog conjugated with a membrane-permeating peptide inhibits human immunodeficiency virus type 1 production. J Virol 2002, 76, 3881–3891. [Google Scholar]
- Gait, MJ. Peptide-mediated cellular delivery of antisense oligonucleotides and their analogues. Cell Mol Life Sci 2003, 60, 844–853. [Google Scholar]
- Tripathi, S; Chaubey, B; Ganguly, S; Harris, D; Casale, RA; Pandey, VN. Anti-HIV-1 activity of anti-TAR polyamide nucleic acid conjugated with various membrane transducing peptides. Nucleic Acids Res 2005, 33, 4345–4356. [Google Scholar]
- Nielsen, PE. PNA Technology. Mol Biotechnol 2004, 26, 233–248. [Google Scholar]
- Turner, JJ; Ivanova, GD; Verbeure, B; Williams, D; Arzumanov, AA; Abes, S; Lebleu, B; Gait, MJ. Cell-penetrating peptide conjugates of peptide nucleic acids (PNA) as inhibitors of HIV-1 Tat-dependent trans-activation in cells. Nucleic Acids Res 2005, 33, 6837–6849. [Google Scholar]
- Turner, JJ; Arzumanov, AA; Gait, MJ. Synthesis, cellular uptake and HIV-1 Tat-dependent trans-activation inhibition activity of oligonucleotide analogues disulphide-conjugated to cell-penetrating peptides. Nucleic Acids Res 2005, 33, 27–42. [Google Scholar]
- Arzumanov, A; Stetsenko, DA; Malakhov, AD; Reichelt, S; Sorensen, MD; Babu, BR; Wengel, J; Gait, MJ. A structure-activity study of the inhibition of HIV-1 Tat-dependent trans-activation by mixmer 2′-O-methyl oligoribonucleotides containing locked nucleic acid (LNA), alpha-L-LNA, or 2′-thio-LNA residues. Oligonucleotides 2003, 13, 435–453. [Google Scholar]
- Kang, SH; Cho, MJ; Kole, R. Up-regulation of luciferase gene expression with antisense oligonucleotides: implications and applications in functional assay development. Biochemistry 1998, 37, 6235–6239. [Google Scholar]
- Moulton, HM; Nelson, MH; Hatlevig, SA; Reddy, MT; Iversen, PL. Cellular uptake of antisense morpholino oligomers conjugated to arginine-rich peptides. Bioconjug Chem 2004, 15, 290–299. [Google Scholar]
- Neuman, BW; Stein, DA; Kroeker, AD; Paulino, AD; Moulton, HM; Iversen, PL; Buchmeier, MJ. Antisense morpholino-oligomers directed against the 5′ end of the genome inhibit coronavirus proliferation and growth. J Virol 2004, 78, 5891–5899. [Google Scholar]
- Deas, TS; Binduga-Gajewska, I; Tilgner, M; Ren, P; Stein, DA; Moulton, HM; Iversen, PL; Kauffman, EB; Kramer, LD; Shi, PY. Inhibition of flavivirus infections by antisense oligomers specifically suppressing viral translation and RNA replication. J Virol 2005, 79, 4599–4609. [Google Scholar]
- Kinney, RM; Huang, CY; Rose, BC; Kroeker, AD; Dreher, TW; Iversen, PL; Stein, DA. Inhibition of dengue virus serotypes 1 to 4 in vero cell cultures with morpholino oligomers. J Virol 2005, 79, 5116–5128. [Google Scholar]
- Neuman, BW; Stein, DA; Kroeker, AD; Churchill, MJ; Kim, AM; Kuhn, P; Dawson, P; Moulton, HM; Bestwick, RK; Iversen, PL; Buchmeier, MJ. Inhibition, escape, and attenuated growth of severe acute respiratory syndrome coronavirus treated with antisense morpholino oligomers. J Virol 2005, 79, 9665–9676. [Google Scholar]
- Deas, TS; Bennett, CJ; Jones, SA; Tilgner, M; Ren, P; Behr, MJ; Stein, DA; Iversen, PL; Kramer, LD; Bernard, KA; Shi, PY. In vitro resistance selection and in vivo efficacy of morpholino oligomers against West Nile virus. Antimicrob Agents Chemother 2007, 51, 2470–2482. [Google Scholar]
- Lebleu, B; Moulton, HM; Abes, R; Ivanova, GD; Abes, S; Stein, DA; Iversen, PL; Arzumanov, AA; Gait, MJ. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv Drug Deliv Rev 2008, 60, 517–529. [Google Scholar]
- Rothbard, JB; Kreider, E; VanDeusen, CL; Wright, L; Wylie, BL; Wender, PA. Arginine-rich molecular transporters for drug delivery: role of backbone spacing in cellular uptake. J Med Chem 2002, 45, 3612–3618. [Google Scholar]
- Abes, S; Moulton, HM; Clair, P; Prevot, P; Youngblood, DS; Wu, RP; Iversen, PL; Lebleu, B. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J Control Release 2006, 116, 304–313. [Google Scholar]
- Moulton, HM; Fletcher, S; Neuman, BW; McClorey, G; Stein, DA; Abes, S; Wilton, SD; Buchmeier, MJ; Lebleu, B; Iversen, PL. Cell-penetrating peptide-morpholino conjugates alter pre-mRNA splicing of DMD (Duchenne muscular dystrophy) and inhibit murine coronavirus replication in vivo. Biochem Soc Trans 2007, 35, 826–828. [Google Scholar]
- Efimov, V; Choob, M; Buryakova, A; Phelan, D; Chakhmakhcheva, O. PNA-related oligonucleotide mimics and their evaluation for nucleic acid hybridization studies and analysis. Nucleosides Nucleotides Nucleic Acids 2001, 20, 419–428. [Google Scholar]
- Wang, Y; Stricker, HM; Gou, D; Liu, L. MicroRNA: past and present. Front Biosci 2007, 12, 2316–2329. [Google Scholar]
- Krützfeldt, J; Rajewsky, N; Braich, R; Rajeev, KG; Tuschl, T; Manoharan, M; Stoffel, M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature 2005, 438, 685–689. [Google Scholar]
- Fisher, L; Samuelsson, M; Jiang, Y; Ramberg, V; Figueroa, R; Hallberg, E; Langel, Ü; Iverfeldt, K. Targeting cytokine expression in glial cells by cellular delivery of an NF-kappaB decoy. J Mol Neurosci 2007, 31, 209–219. [Google Scholar]
- El-Andaloussi, S; Johansson, H; Magnusdottir, A; Järver, P; Lundberg, P; Langel, Ü. TP10, a delivery vector for decoy oligonucleotides targeting the Myc protein. J Control Release 2005, 110, 189–201. [Google Scholar]
- Nagatsugi, F; Sasaki, S. Chemical tools for targeted mutagenesis of DNA based on triple helix formation. Biol Pharm Bull 2004, 27, 463–467. [Google Scholar]
- Rogers, FA; Manoharan, M; Rabinovitch, P; Ward, DC; Glazer, PM. Peptide conjugates for chromosomal gene targeting by triplex-forming oligonucleotides. Nucleic Acids Res 2004, 32, 6595–6604. [Google Scholar]
- Ding, SW (Ed.) RNAi: Mechanisms, Biology and Applications. FEBS Lett 2005, 579, 5821–6007.
- Rana, TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol 2007, 8, 23–36. [Google Scholar]
- Davidson, TJ; Harel, S; Arboleda, VA; Prunell, GF; Shelanski, ML; Greene, LA; Troy, CM. Highly efficient small interfering RNA delivery to primary mammalian neurons induces MicroRNA-like effects before mRNA degradation. J Neurosci 2004, 24, 10040–10046. [Google Scholar]
- Meade, BR; Dowdy, SF. Enhancing the cellular uptake of siRNA duplexes following noncovalent packaging with protein transduction domain peptides. Adv Drug Deliv Rev 2007. [Google Scholar]
- Tönges, L; Lingor, P; Egle, R; Dietz, GP; Fahr, A; Bahr, M. Stearylated octaarginine and artificial virus-like particles for transfection of siRNA into primary rat neurons. RNA 2006, 12, 1431–1438. [Google Scholar]
- Moschos, SA; Williams, AE; Lindsay, MA. Cell-penetrating-peptide-mediated siRNA lung delivery. Biochem Soc Trans 2007, 35, 807–810. [Google Scholar]
- Kumar, P; Wu, H; McBride, JL; Jung, KE; Kim, MH; Davidson, BL; Lee, SK; Shankar, P; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar]
- Spagnou, S; Miller, AD; Keller, M. Lipidic carriers of siRNA: differences in the formulation, cellular uptake, and delivery with plasmid DNA. Biochemistry 2004, 43, 13348–13356. [Google Scholar]
- Veldhoen, S; Laufer, SD; Trampe, A; Restle, T. Cellular delivery of small interfering RNA by a non-covalently attached cell-penetrating peptide: quantitative analysis of uptake and biological effect. Nucleic Acids Res 2006, 34, 6561–6573. [Google Scholar]
- Mescalchin, A; Detzer, A; Wecke, M; Overhoff, M; Wünsche, W; Sczakiel, G. Cellular uptake and intracellular release are major obstacles to the therapeutic application of siRNA: novel options by phosphorothioate-stimulated delivery. Expert Opin Biol Ther 2007, 7, 1531–1538. [Google Scholar]
- Overhoff, M; Wünsche, W; Sczakiel, G. Quantitative detection of siRNA and single-stranded oligonucleotides: relationship between uptake and biological activity of siRNA. Nucleic Acids Res 2004, 32, e170. [Google Scholar]
- Deshayes, S; Morris, MC; Divita, G; Heitz, F. Interactions of amphipathic CPPs with model membranes. Biochim Biophys Acta 2006, 1758, 328–335. [Google Scholar]

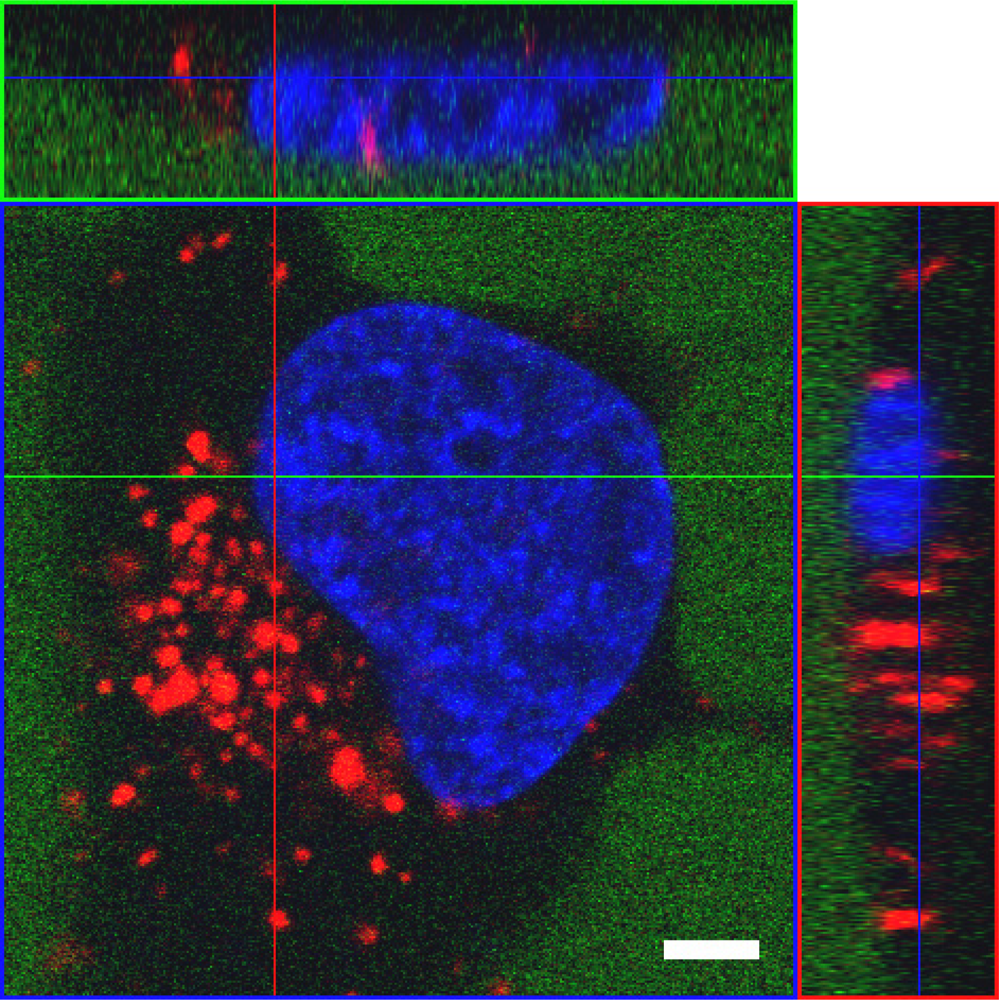

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence | Reference |
|---|---|---|
| Tat48–60 | GRKKRRQRRRPPQ | [20] |
| penetratin (Antp43–58) | RQIKIWFQNRRMKWKK | [63] |
| transportan | GWTLNSAGYLLGKINLKALAALAKKIL | [64] |
| TP10 | AGYLLGKINLKALAALAKKIL | [65] |
| Oligoarginine (R8) | RRRRRRRR | [22,66] |
| MAP | KLALKLALKALKAALKLA | [67] |
| MPG | GALFLGFLGAAGSTMGAWSQPKKKRKV | [19] |
| MPGα | GALFLAFLAAALSLMGLWSQPKKKRKV | [68] |
| Peptide | Sequence | Reference |
|---|---|---|
| hCT9–32-br | LGTYTQDFNK*FHTFPQTAIGVGAP (-AFGVGPDEVKRKKKP; attached to K*) | [69] |
| SAP | (VRLPPP)3 | [70] |
| S413-PV | ALWKTLLKKVLKAPKKKRKV | [71] |
| mPrPp | MANLGYWLLALFVTMWTDVGLCKKRPKP | [72,73] |
| bPrPp | MVKSKIGSWILVLFVAMWSDVGLCKKRPKP | [73,74] |
| M918 | MVTVLFRRLRIRRACGPPRVRV | [54] |
| CPP5s | VPMLK, PMLKE (human), VPTLK (mouse), VPALR (rat) | [75] |
| EB1 | LIRLWSHLIHIWFQNRRLKWKKK | [76] |
| Cargo | CPP/delivery system | proposed uptake mechanism | Reference |
|---|---|---|---|
| plasmid DNA | MPG | non-endocytotic | [130] |
| plasmid DNA | R8, stearyl-R8, Tat48–60 | endocytotic | [197] |
| plasmid DNA | branched 8Tat peptide | endocytotic | [195] |
| plasmid DNA | Tat48–60-peptide | endocytotic | [91] |
| plasmid DNA | Tat47–57-oligomers | endocytotic | [93] |
| plasmid DNA | Tat47–57 | endocytotic | [92] |
| plasmid DNA | branched 8Tat peptide | endocytotic (dependent on cell line) | [196] |
| plasmid DNA | POLYTAT | endocytotic | [193] |
| Plasmid | bPrPp | endocytotic | [161] |
| DNA/looped DNA | |||
| plasmid DNA | hCT9–32-2br, hCT18–32-k7 | endocytotic | [142] |
| plasmid DNA | R-PAMAM-PEG-PAMAM-R dendrimer | endocytotic | [198] |
| plasmid DNA | HK-polymer | endocytotic | [183,184] |
| plasmid DNA | R8-MEND3 | endocytotic | [179] |
| Cargo | CPP/delivery system | proposed uptake mechanism | Reference |
|---|---|---|---|
| DNA oligonucleotide | MPG | non-endocytotic | [19] |
| antisense PNA | penetratin, transportan | n. d. | [112] |
| antisense PNA | transportan | n. d. | [203] |
| antisense PNA | Tat48–60, penetratin, transportan analogues | non-endocytotic | [205] |
| antisense PNA | Tat48–58, penetratin, transportan analogues, R9F2, R6-penetratin and further peptides | endocytotic (transportan: possibly non-endocytotic) | [207] |
| antisense PMO | R9F2 (non-covalent + covalent), Tat peptide, penetratin (covalent) | n. d. | [211] |
| antisense PMO | (R-Ahx-R)4 | endocytotic | [219] |
| antisense PNA | R6-penetratin | endocytotic | [109] |
| antisense PMO | (R-Ahx-R)4AhxB | n. d. | [220] |
| HypNA-pPNA | Pep-3 | n. d. (proposed non-endocytotic) | [134] |
| Antagomir | R6-penetratin | n. d. | [108] |
| antisense 2'-OMe phosphorothioate RNA oligonucleotides | Tat49–60, penetratin | endocytotic | [87] |
| antisense RNA oligonucleotide analogues | Tat48–58, penetratin, R6-penetratin, transportan, R9, R9F2 and further peptides | endocytotic | [208] |
| decoy | PNA-coupled transportan or TP10 | n. d. | [113,224] |
| decoy | PNA-coupled transportan or TP10 + NLS | non-endocytotic, to small extent endocytotic | [225] |
| TFO | penetratin | non-endocytotic endocytotic pathway not excluded | [227] |
| Cargo | CPP/delivery system | proposed uptake mechanism | Reference |
|---|---|---|---|
| siRNA | MPG, MPGΔNLS (non-covalent) | non-endocytotic | [131] |
| siRNA | penetratin, transportan | n. d. | [104] |
| siRNA | Tat47–57, Tat-derived oligocarbamate | n. d. | [90] |
| siRNA | penetratin | n. d. | [230] |
| siRNA | H3K8b, H3K8b(+RGD) | endocytotic | [172] |
| siRNA | stearyl-R8 | endocytotic | [232] |
| siRNA | Chol-R9 | endocytotic | [121] |
| siRNA | MPGα | endocytotic | [236] |
| siRNA | R8-MEND (siRNA/stearyl-R8core) | endocytotic | [187] |
| siRNA | EB1, MPGΔNLS, bPrPp | endocytotic | [76] |
| siRNA | Tat48–60, penetratin | endocytotic | [106] |
| siRNA | RVG peptide | n. d. | [234] |
| siRNA | rCPP | endocytotic | [181] |
Share and Cite
Veldhoen, S.; Laufer, S.D.; Restle, T. Recent Developments in Peptide-Based Nucleic Acid Delivery. Int. J. Mol. Sci. 2008, 9, 1276-1320. https://doi.org/10.3390/ijms9071276
Veldhoen S, Laufer SD, Restle T. Recent Developments in Peptide-Based Nucleic Acid Delivery. International Journal of Molecular Sciences. 2008; 9(7):1276-1320. https://doi.org/10.3390/ijms9071276
Chicago/Turabian StyleVeldhoen, Sandra, Sandra D. Laufer, and Tobias Restle. 2008. "Recent Developments in Peptide-Based Nucleic Acid Delivery" International Journal of Molecular Sciences 9, no. 7: 1276-1320. https://doi.org/10.3390/ijms9071276
APA StyleVeldhoen, S., Laufer, S. D., & Restle, T. (2008). Recent Developments in Peptide-Based Nucleic Acid Delivery. International Journal of Molecular Sciences, 9(7), 1276-1320. https://doi.org/10.3390/ijms9071276