Key Labeling Technologies to Tackle Sizeable Problems in RNA Structural Biology

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
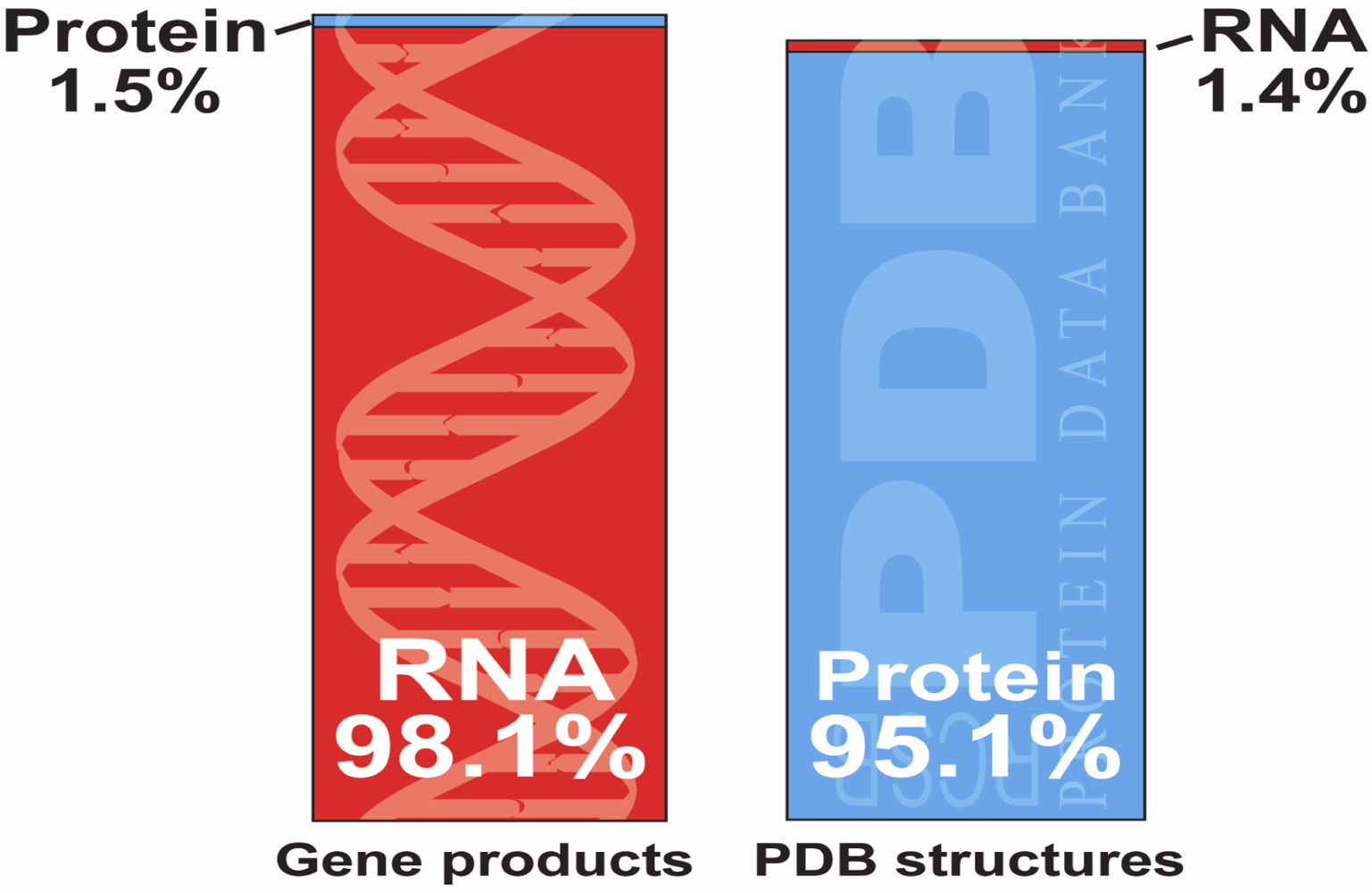
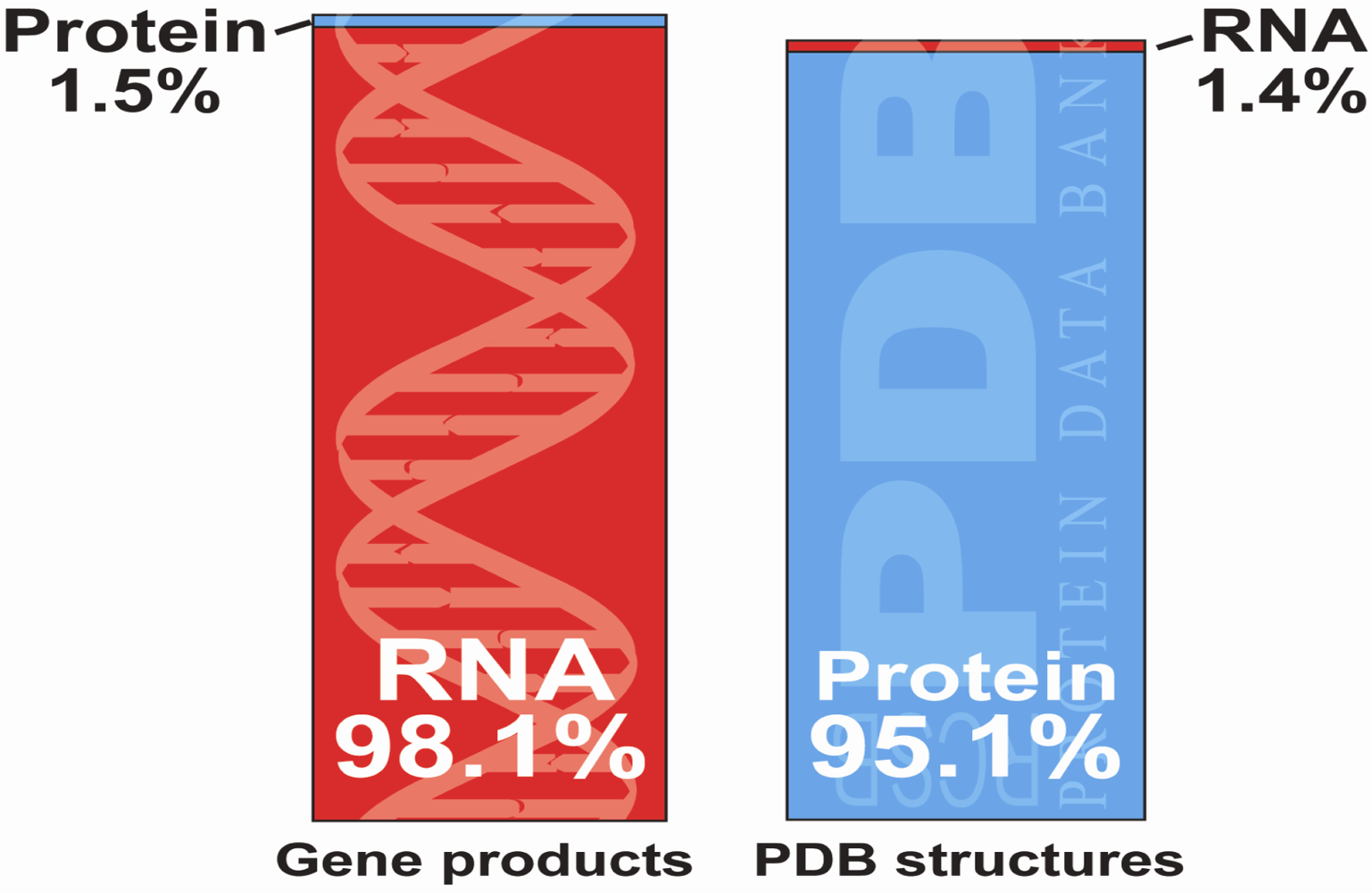
:1. Introduction
2. Synthesis and purification of milligram quantities of natively folded RNA for biophysical studies
2.1 T7 RNA polymerase based transcription of RNA
2.2 Native purification of RNA for biophysical studies
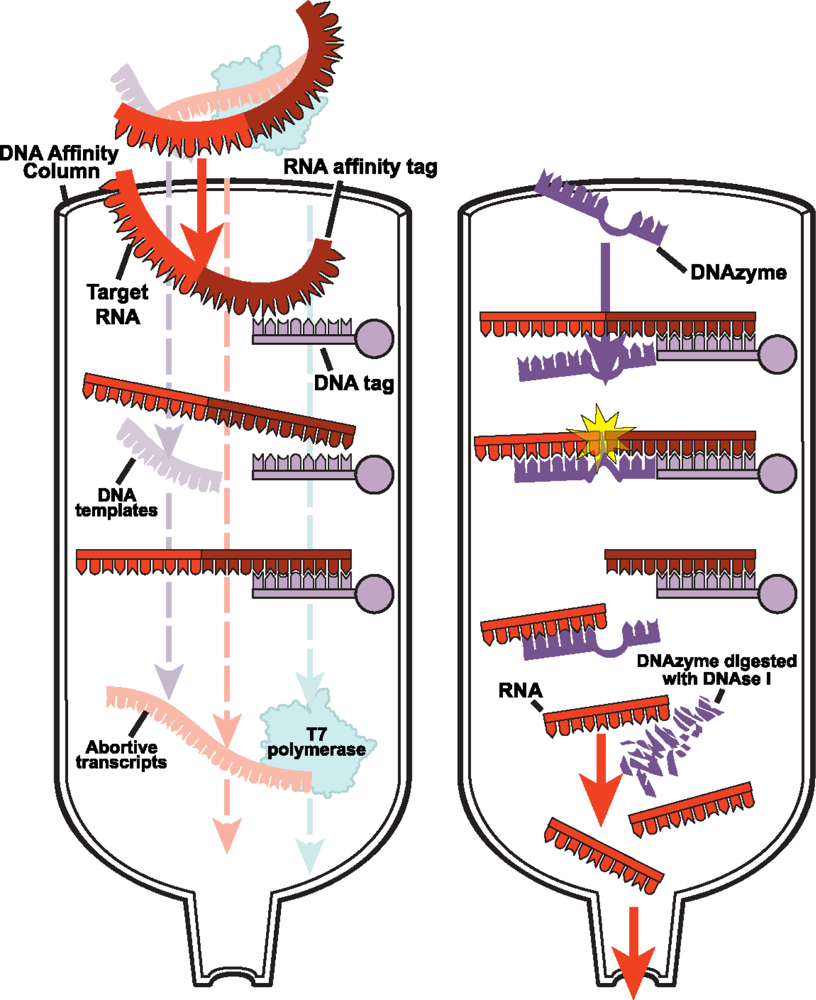
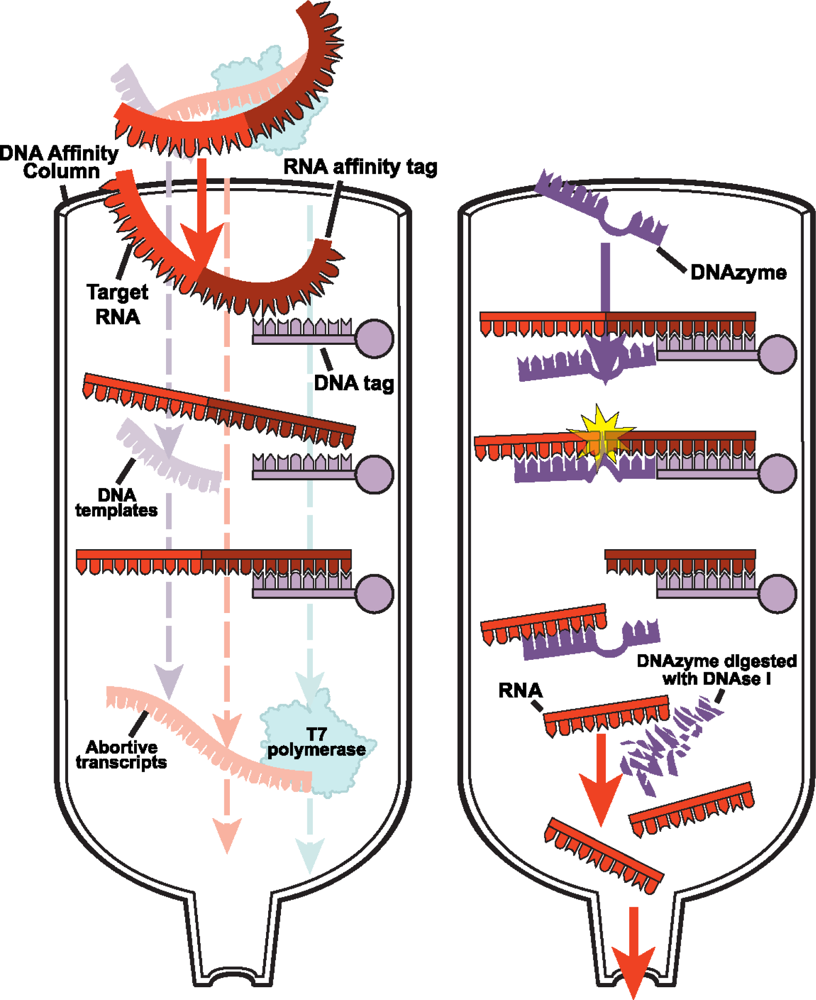
2.2.1 DNA affinity column chromatography for native purification of RNA
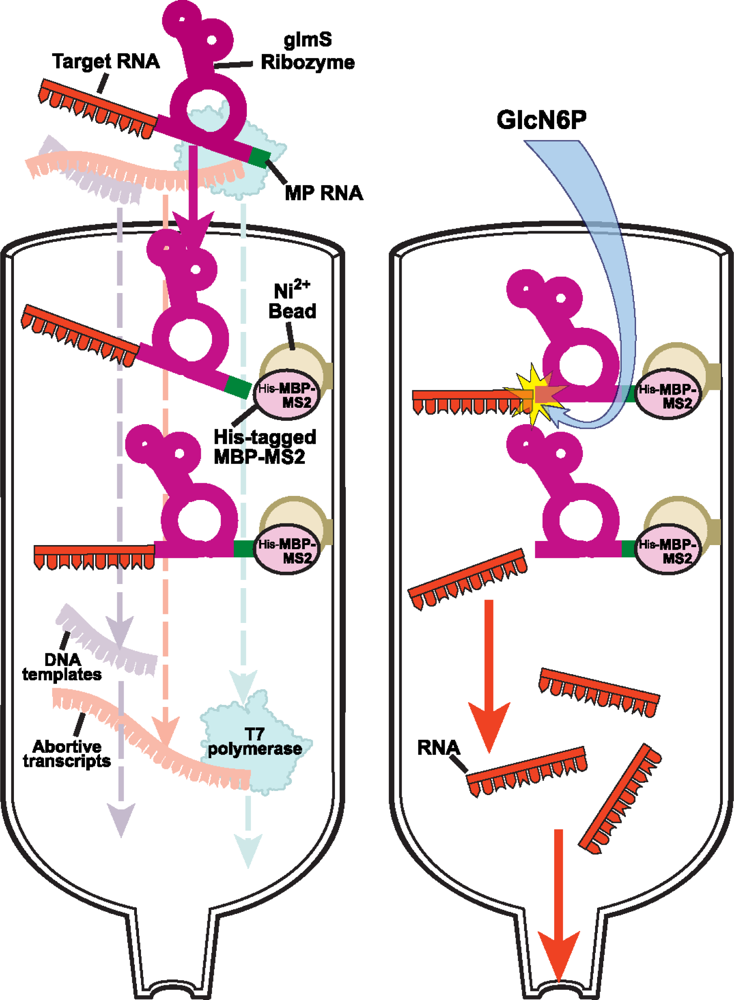
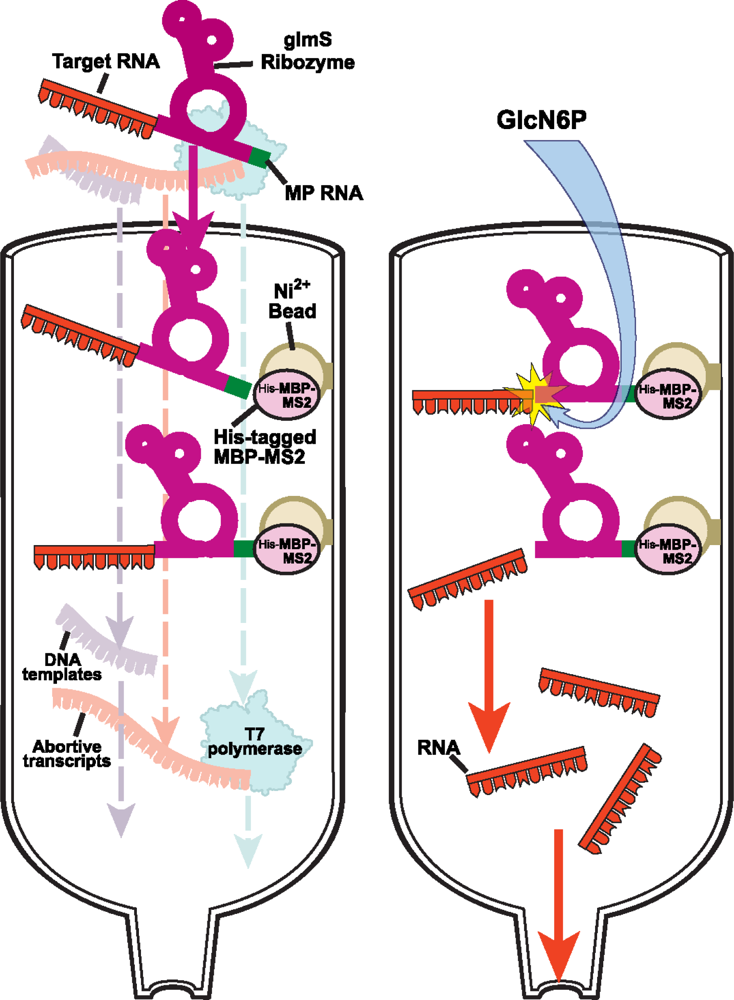
2.2.2 Protein-RNA affinity column chromatography for native purification of RNA
2.2.3 Size exclusion chromatography for native purification of RNA
3. Isotopic labeling of RNA for biophysical studies
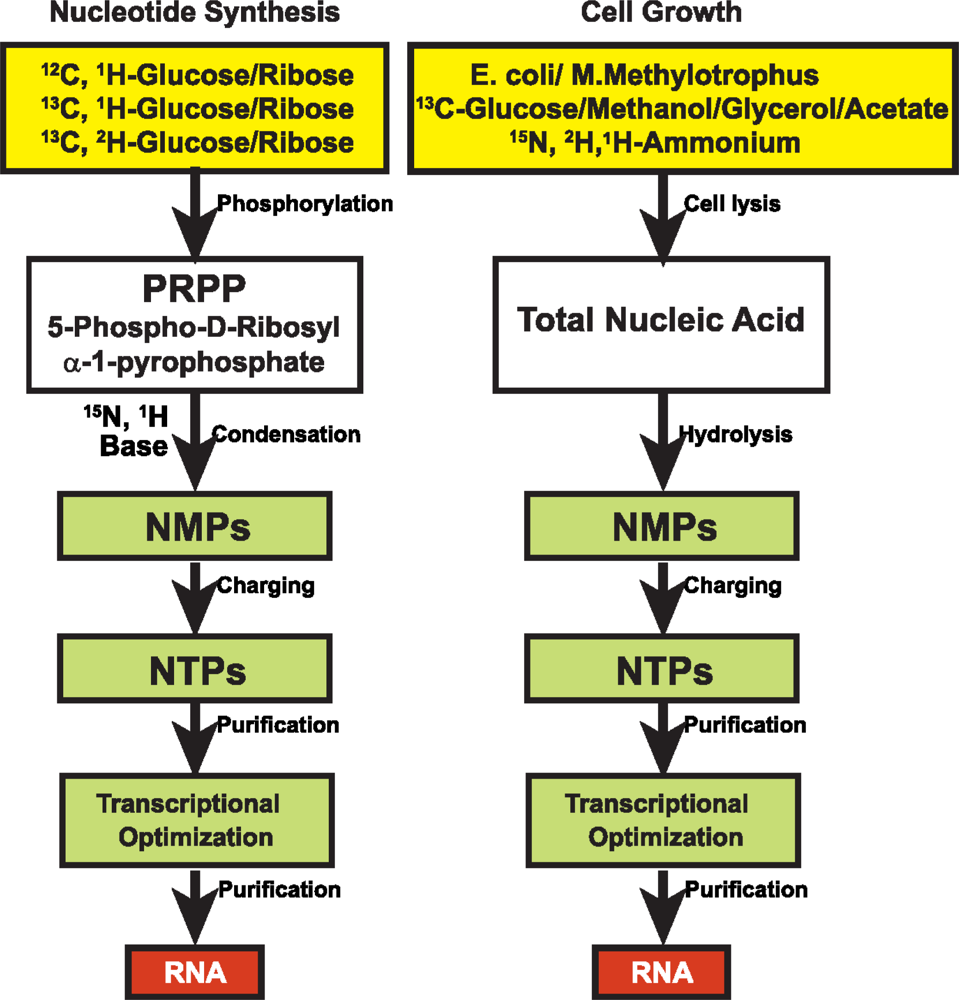
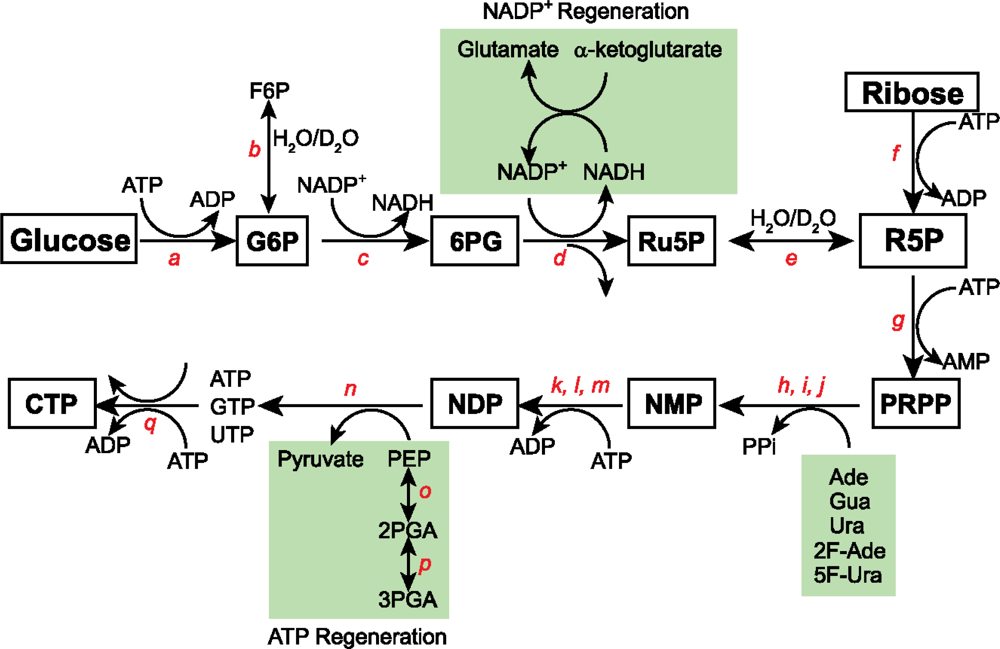
3.1 15N and 13C isotopic labeling of RNA for biophysical studies
3.2 Alternate site-specific 13C isotopic labeling of RNA with different E.coli strains
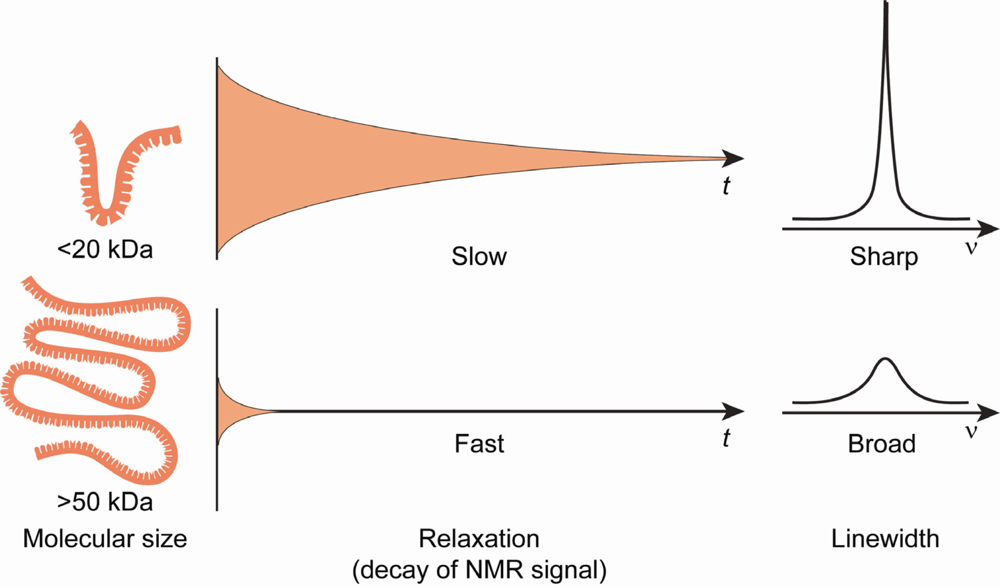
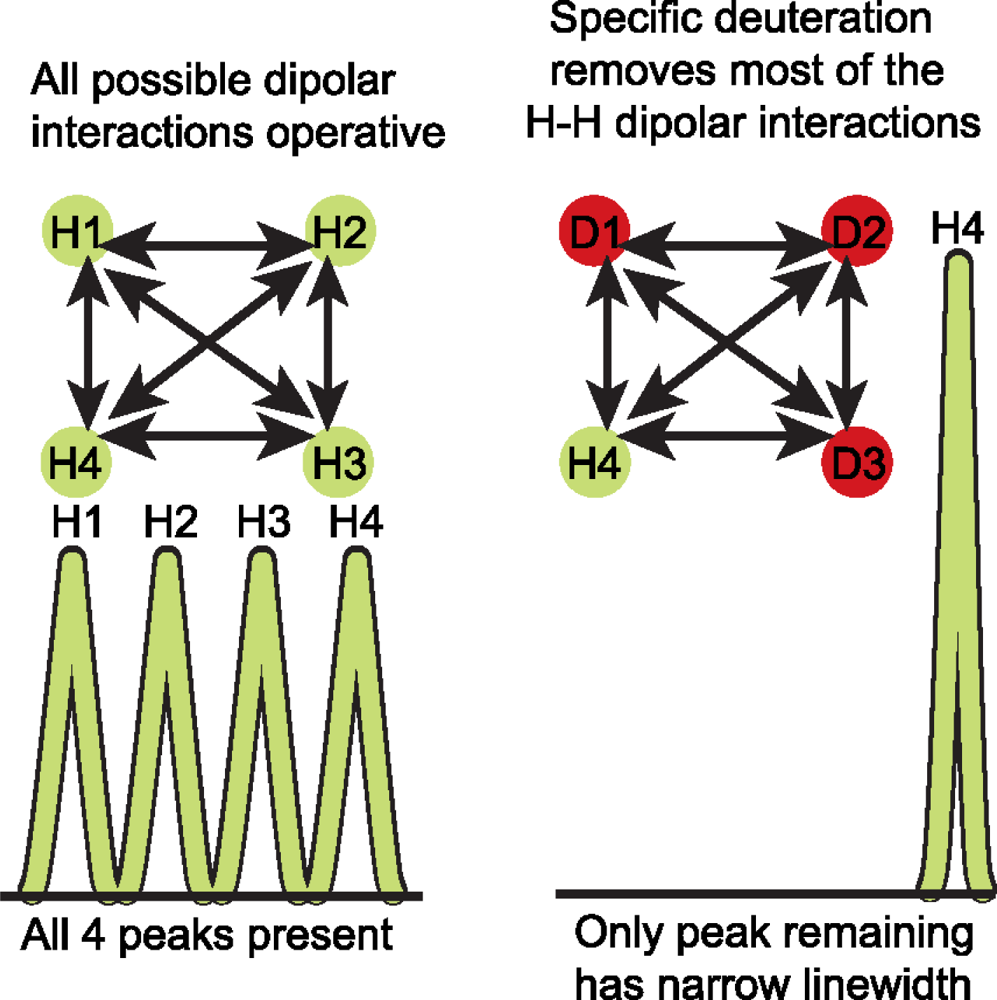
3.3 Deuterium (2H) isotopic labeling of RNA for biophysical studies
3.4 2H, 13C, and 19F isotopic labeling of RNA for biophysical studies
3.5 Phasing strategies in RNA crystallography
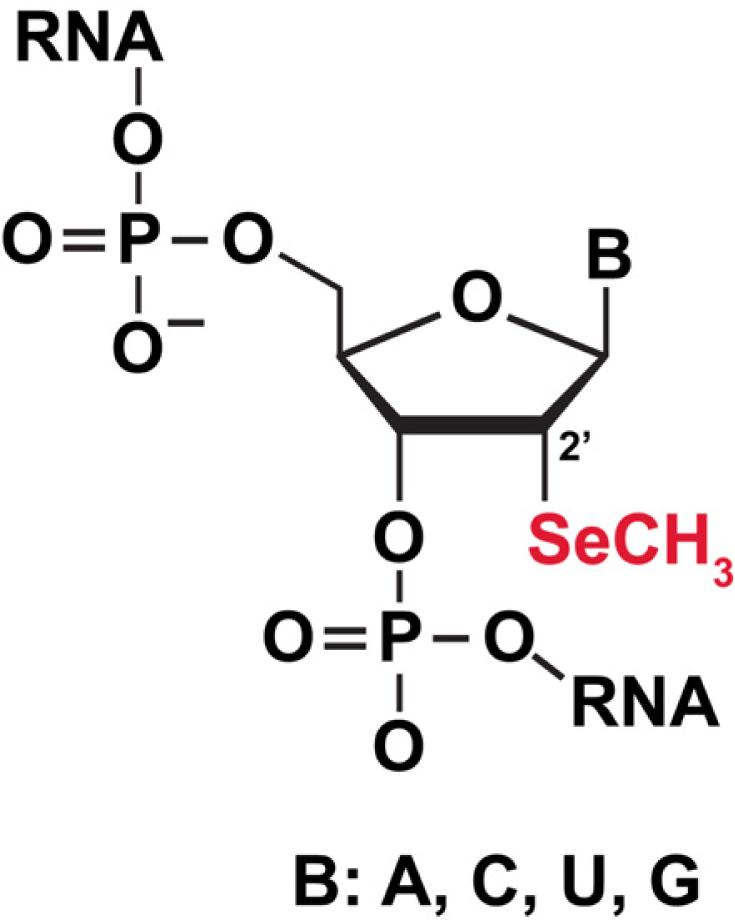
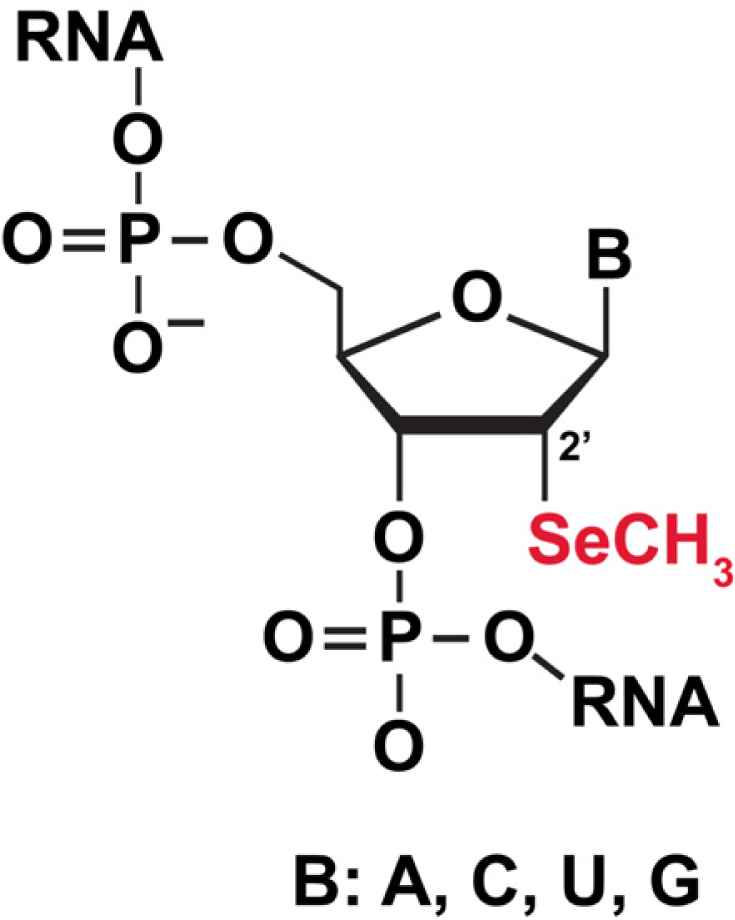
3.5.1 Selenium (Se) labeling of RNA for biophysical studies
3.5.2 GU platforms for direct soaking of divalent metals for RNA for biophysical studies
4. Improved methods for ligating large RNAs for biophysical studies
4.1 Chemical ligation
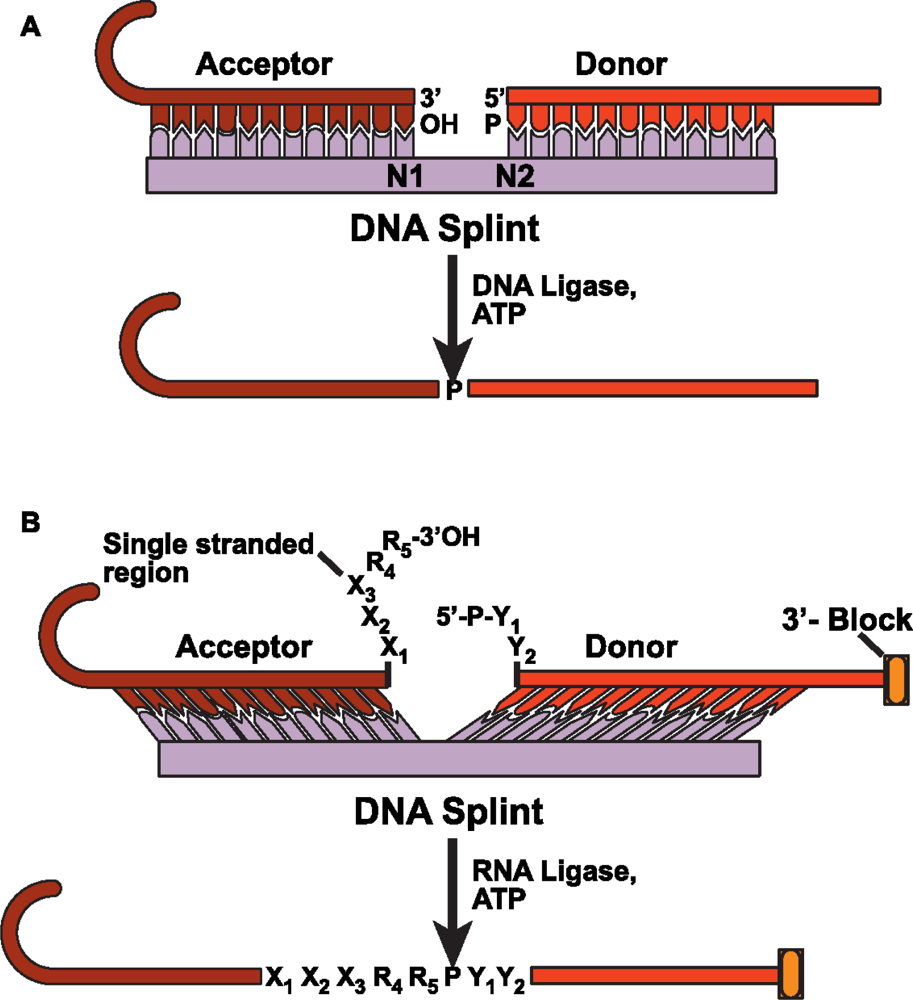
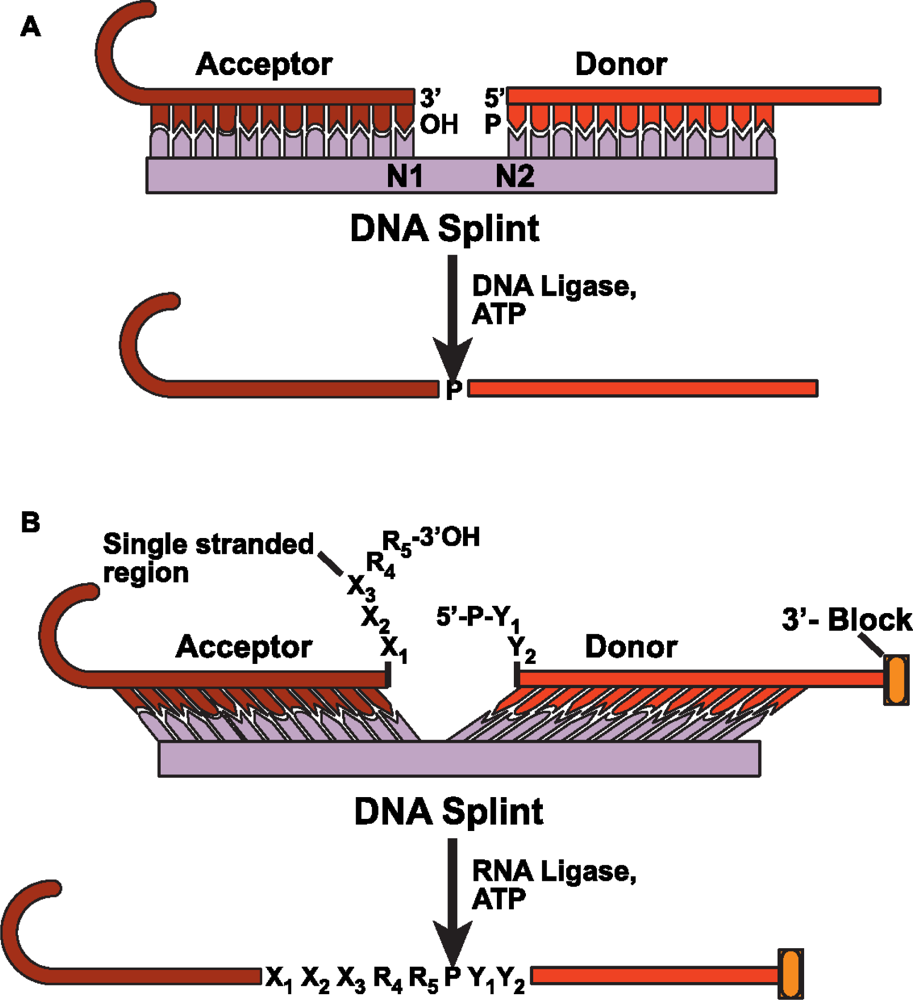
4.2 DNA splinted T4 DNA ligase based ligation
4.3 DNA splinted T4 RNA based ligation
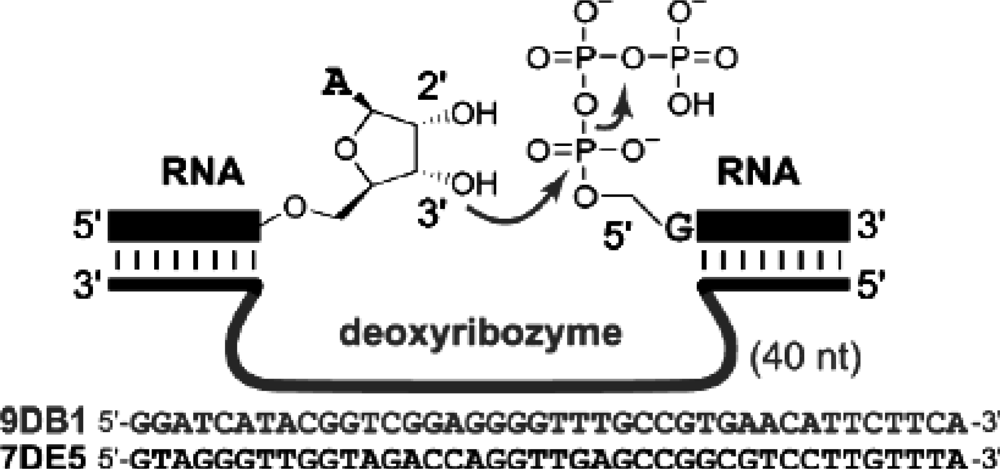
4.4 Deoxyribozyme based ligation
5. Tackling sizeable problems in RNA biophysical chemistry
5.1 Structural basis for encapsidation of the genome of the Moloney Murine Leukemia Virus
5.2 Towards the structural basis for Group II Intron ribozyme catalytic funtion
Acknowledgements
References and Notes
- Gesteland, RF; Cech, TR; Atkins, JF (Eds.) RNA World; Cold Spring Harbor Laboratory Press: New York, 2006; pp. 1–768.
- Korostelev, A; Noller, HF. The ribosome in focus: new structures bring new insights. Trends Biochem Sci 2007, 32, 434–441. [Google Scholar]
- Steitz, TA. A structural understanding of the dynamic ribosome machine. Nat Rev Mol Cell Biol 2008, 9, 242–253. [Google Scholar]
- Stark, H; Lührmann, R. Cryo-electron microscopy of spliceosomal components. Annu Rev Biophys Biomol Struct 2006, 35, 435–457. [Google Scholar]
- Bessonov, S; Anokhina, M; Will, CL; Urlaub, H; Luhrmann, R. Isolation of an active step I spliceosome and composition of its RNP core. Nature 2008, 452, 846–850. [Google Scholar]
- Boisvert, FM; van Koningsbruggen, S; Navascués, J; Lamond, AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol 2007, 8, 574–585. [Google Scholar]
- Wakeman, CA; Winkler, WC; Dann, CE, 3rd. Structural features of metabolite-sensing riboswitches. Trends Biochem Sci 2007, 32, 415–424. [Google Scholar]
- Edwards, TE; Klein, DJ; Ferré-D'Amaré, AR. Riboswitches: small-molecule recognition by gene regulatory RNAs. Curr Opin Struct Biol 2007, 17, 273–279. [Google Scholar]
- Taft, RJ; Pheasant, M; Mattick, JS. The relationship between non-protein-coding DNA and eukaryotic complexity. Bioessays 2007, 29, 288–99. [Google Scholar]
- Amaral, PP; Dinger, ME; Mercer, TR; Mattick, JS. The eukaryotic genome as an RNA machine. Science 2008, 319, 1787–1789. [Google Scholar]
- Cromsigt, J; van Buuren, B; Schleucher, J; Wijmenga, S. Resonance assignment and structure determination for RNA. Methods Enzymol 2001, 338, 371–399. [Google Scholar]
- Wijmenga, SS; van Buuren, BNM. The use of NMR methods for conformational studies of nucleic acids. Prog NMR Spectrosc 1998, 32, 287–387. [Google Scholar]
- Furtig, B; Richter, C; Bermel, W; Schwalbe, H. New NMR experiments for RNA nucleobase resonance assignment and chemical shift analysis of an RNA UUCG tetraloop. J Biomol NMR 2004, 28, 69–79. [Google Scholar]
- Latham, MP; Brown, DJ; McCallum, SA; Pardi, A. NMR methods for studying the structure and dynamics of RNA. Chembiochem 2005, 6, 1492–1505. [Google Scholar]
- Tzakos, AG; Grace, CR; Lukavsky, PJ; Riek, R. NMR techniques for very large proteins and RNAs in solution. Annu Rev Biophys Biomol Struct 2006, 35, 319–342. [Google Scholar]
- Golden, BL; Kundrot, CE. RNA crystallization. J Struct Biol 2003, 142, 98–107. [Google Scholar]
- Golden, BL. Preparation and crystallization of RNA. Methods Mol Biol 2007, 363, 239–257. [Google Scholar]
- Cate, JH; Doudna, JA. Solving large RNA structures by X-ray crystallography. Methods Enzymol 2000, 317, 169–180. [Google Scholar]
- Ke, A; Doudna, JA. Crystallization of RNA and RNA-protein complexes. Methods 2004, 34, 408–414. [Google Scholar]
- Scaringe, SA. RNA oligonucleotide synthesis via 5′-silyl-2′-orthoester chemistry. Methods 2001, 23, 206–217. [Google Scholar]
- Krieg, PA; Melton, DA. In vitro RNA synthesis with SP6 RNA polymerase. Methods Enzymol 1987, 155, 397–415. [Google Scholar]
- Milligan, JF; Uhlenbeck, OC. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol 1989, 180, 51–62. [Google Scholar]
- Pokrovskaya, ID; Gurevich, VV. In vitro transcription: preparative RNA yields in analytical scale reactions. Anal Biochem 1994, 220, 420–423. [Google Scholar]
- Studier, FW; Rosenberg, AH; Dunn, JJ; Dubendorff, JW. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol 1990, 185, 60–89. [Google Scholar]
- Pleiss, JA; Derrick, ML; Uhlenbeck, OC. T7 RNA polymerase produces 5′ end heterogeneity during in vitro transcription from certain templates. RNA 1998, 4, 1313–1317. [Google Scholar]
- Helm, M; Brulé, H; Giegé, R; Florentz, C. More mistakes by T7 RNA polymerase at the 5′ ends of in vitro-transcribed RNAs. RNA 1999, 5, 618–621. [Google Scholar]
- Krupp, G. RNA synthesis: strategies for the use of bacteriophage RNA polymerases. Gene 1988, 72, 75–89. [Google Scholar]
- Wyatt, JR; Chastain, M; Puglisi, JD. Synthesis and purification of large amounts of RNA oligonucleotides. Biotechniques 1991, 11, 764–769. [Google Scholar]
- Puglisi, JD; Wyatt, JR. Biochemical and NMR studies of RNA conformation with an emphasis on RNA pseudoknots. Methods Enzymol 1995, 261, 323–350. [Google Scholar]
- Grosshans, CA; Cech, TR. A hammerhead ribozyme allows synthesis of a new form of the Tetrahymena ribozyme homogeneous in length with a 3′ end blocked for transesterification. Nucleic Acids Res 1991, 19, 3875–3880. [Google Scholar]
- Ferré-D'Amaré, AR; Doudna, JA. Use of cis- and trans-ribozymes to remove 5′ and 3′ heterogeneities from milligrams of in vitro transcribed RNA. Nucleic Acids Res 1996, 24, 977–978. [Google Scholar]
- Moran, S; Ren, RX; Sheils, CJ; Rumney, S, 4th; Kool, ET, 4th. Non-hydrogen bonding ‘terminator’ nucleosides increase the 3′-end homogeneity of enzymatic RNA and DNA synthesis. Nucleic Acids Res 1996, 24, 2044–2052. [Google Scholar]
- Kao, C; Zheng, M; Rüdisser, S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. RNA 1999, 5, 1268–1272. [Google Scholar]
- Coleman, TM; Wang, G; Huang, F. Superior 5′ homogeneity of RNA from ATP-initiated transcription under the T7 phi 2.5 promoter. Nucleic Acids Res 2004, 32, e14. [Google Scholar]
- Dayie, KT. Resolution enhanced homonuclear carbon decoupled triple resonance experiments for unambiguous RNA structural characterization. J Biomol NMR 2005, 32, 129–139. [Google Scholar]
- Ponchon, L; Dardel, F. Recombinant RNA technology: the tRNA scaffold. Nat Methods 2007, 4, 571–576. [Google Scholar] [Green Version]
- Wincott, F; DiRenzo, A; Shaffer, C; Grimm, S; Tracz, D; Workman, C; Sweedler, D; Gonzalez, C; Scaringe, S; Usman, N. Synthesis, deprotection, analysis and purification of RNA and ribozymes. Nucleic Acids Res 1995, 23, 2677–2684. [Google Scholar]
- Anderson, AC; Scaringe, SA; Earp, BE; Frederick, CA. HPLC purification of RNA for crystallography and NMR. RNA 1996, 2, 110–117. [Google Scholar]
- Shields, TP; Mollova, E; Ste Marie, L; Hansen, MR; Pardi, A. High-performance liquid chromatography purification of homogenous-length RNA produced by trans cleavage with a hammerhead ribozyme. RNA 1999, 5, 1259–1267. [Google Scholar]
- Sich, C; Ohlenschlager, O; Ramachandran, R; Gorlach, M; Brown, LR. Structure of an RNA hairpin loop with a 5′-CGUUUCG-3′ loop motif by heteronuclear NMR spectroscopy and distance geometry. Biochemistry 1997, 36, 13989–4002. [Google Scholar]
- Azarani, A; Hecker, KH. RNA analysis by ion-pair reversed-phase high performance liquid chromatography. Nucleic Acids Res 2001, 29, e7. [Google Scholar]
- Dickman, MJ; Conroy, MJ; Grasby, JA; Hornby, DP. RNA footprinting analysis using ion pair reverse phase liquid chromatography. RNA 2002, 8, 247–251. [Google Scholar]
- Nikonowicz, EP; Sirr, A; Legault, P; Jucker, FM; Baer, LM; Pardi, A. Preparation of 13C and 15N labelled RNAs for heteronuclear multi-dimensional NMR studies. Nucleic Acids Res 1992, 20, 4507–4513. [Google Scholar]
- Batey, RT; Inada, M; Kujawinski, E; Puglisi, JD; Williamson, JR. Preparation of isotopically labeled ribonucleotides for multidimensional NMR spectroscopy of RNA. Nucleic Acids Res 1992, 20, 4515–4523. [Google Scholar]
- Cheong, HK; Hwang, E; Lee, C; Choi, BS; Cheong, C. Rapid preparation of RNA samples for NMR spectroscopy and X-ray crystallography. Nucleic Acids Res 2004, 32, e84. [Google Scholar]
- Kieft, JS; Batey, RT. A general method for rapid and nondenaturing purification of RNAs. RNA 2004, 10, 988–995. [Google Scholar]
- Batey, RT; Kieft, JS. Improved native affinity purification of RNA. RNA 2007, 13, 1384–1389. [Google Scholar]
- Crowe, J; Döbeli, H; Gentz, R; Hochuli, E; Stüber, D; Henco, K. 6xHis-Ni-NTA chromatography as a superior technique in recombinant protein expression/purification. Methods Mol Biol 1994, 31, 371–387. [Google Scholar]
- Winkler, WC; Nahvi, A; Roth, A; Collins, JA; Breaker, RR. Control of gene expression by a natural metabolite-responsive ribozyme. Nature 2004, 428, 281–286. [Google Scholar]
- Hampel, KJ; Tinsley, MM. Evidence for preorganization of the glmS ribozyme ligand binding pocket. Biochemistry 2006, 45, 7861–7871. [Google Scholar]
- Lukavsky, PJ; Puglisi, JD. Large-scale preparation and purification of polyacrylamide-free RNA oligonucleotides. RNA 2004, 10, 889–893. [Google Scholar]
- Kim, I; McKenna, SA; Viani-Puglisi, E; Puglisi, JD. Rapid purification of RNAs using fast performance liquid chromatography (FPLC). RNA 2007, 13, 289–294. [Google Scholar]
- McKenna, SA; Kim, I; Puglisi, EV; Lindhout, DA; Aitken, CE; Marshall, RA; Puglisi, JD. Purification and characterization of transcribed RNAs using gel filtration chromatography. Nat Protoc 2007, 2, 3270–3277. [Google Scholar]
- Michnicka, MJ; Harper, JW; King, GC. Selective isotopic enrichment of synthetic RNA: application to the HIV-1 TAR element. Biochemistry 1993, 32, 395–400. [Google Scholar]
- Hines, JV; Landry, SM; Varani, G; Tinoco, I. Carbon-Proton Scalar Couplings in RNA: 3D Heteronuclear and 2D Isotope-Edited NMR of a 13C-Labeled Extra-stable Hairpin. J Am Chem Soc 1994, 116, 5823–5831. [Google Scholar]
- McIntosh, LP; Dahlquist, FW. Biosynthetic incorporation of 15N and 13C for assignment and interpretation of nuclear magnetic resonance spectra of proteins. Q Rev Biophys 1990, 23, 1–38. [Google Scholar]
- Dieckmann, T; Feigon, J. Assignment methodology for larger RNA oligonucleotides: application to an ATP-binding RNA aptamer. J Biomol NMR 1997, 9, 259–272. [Google Scholar]
- Pardi, A. Multidimensional heteronuclear NMR experiments for structure determination of isotopically labeled RNA. Methods Enzymol 1995, 261, 350–380. [Google Scholar]
- Latham, MP; Brown, DJ; McCallum, SA; Pardi, A. NMR methods for studying the structure and dynamics of RNA. Chembiochem 2005, 6, 1492–1505. [Google Scholar]
- Batey, RT; Cloutier, N; Mao, H; Williamson, JR. Improved large scale culture of Methylophilus methylotrophus for 13C/15N labeling and random fractional deuteration of ribonucleotides. Nucleic Acids Res 1996, 24, 4836–4837. [Google Scholar]
- D'Souza, V; Dey, A; Habib, D; Summers, MF. NMR structure of the 101-nucleotide core encapsidation signal of the Moloney murine leukemia virus. J Mol Biol 2004, 337, 427–442. [Google Scholar]
- Peterson, RD; Theimer, CA; Wu, H; Feigon, J. New applications of 2D filtered/edited NOESY for assignment and structure elucidation of RNA and RNA-protein complexes. J Biomol NMR 2004, 28, 59–67. [Google Scholar]
- Hoffman, DW; Holland, JA. Preparation of carbon-13 labeled ribonucleotides using acetate as an isotope source. Nucleic Acids Res 1995, 23, 3361–3362. [Google Scholar]
- Johnson, JE, Jr; Julien, KR; Hoogstraten, CG. Alternate-site isotopic labeling of ribonucleotides for NMR studies of ribose conformational dynamics in RNA. J Biomol NMR 2006, 35, 261–274. [Google Scholar]
- Hoogstraten, CG; Johnson, JE, Jr. Metabolic labeling: Taking advantage of bacterial pathways to prepare spectroscopically useful isotope patterns in proteins and nucleic acids. Concepts in Magnetic Resonance Part A 2008, 32A, 34–55. [Google Scholar]
- Brutscher, B; Boisbouvier, J; Kupce, E; Tisné, C; Dardel, F; Marion, D; Simorre, JP. Base-type-selective high-resolution 13C edited NOESY for sequential assignment of large RNAs. J Biomol NMR 2001, 19, 141–151. [Google Scholar]
- Markley, JL; Putter, I; Jardetzky, O. High-resolution nuclear magnetic resonance spectra of selectively deuterated staphylococcal nuclease. Science 1968, 161, 1249–1251. [Google Scholar]
- LeMaster, DM. Deuterium labelling in NMR structural analysis of larger proteins. Q Rev Biophys 1990, 23, 133–174. [Google Scholar]
- Markus, MA; Dayie, KT; Matsudaira, P; Wagner, G. Effect of deuteration on the amide proton relaxation rates in proteins. Heteronuclear NMR experiments on villin 14T. J Magn Reson B 1994, 105, 192–195. [Google Scholar]
- Tolbert, TJ; Williamson, JR. Preparation of Specifically Deuterated and 13C-Labeled RNA for NMR Studies Using Enzymatic Synthesis. J Am Chem Soc 1997, 119, 12100–12108. [Google Scholar]
- Dayie, KT; Tolbert, TJ; Williamson, JR. 3D C(CC)H TOCSY experiment for assigning protons and carbons in uniformly 13C- and selectively 2H-labeled RNA. J Magn Reson 1998, 130, 97–101. [Google Scholar]
- Gardner, KH; Kay, LE. The use of 2H, 13C, 15N multidimensional NMR to study the structure and dynamics of proteins. Annu Rev Biophys Biomol Struct 1998, 27, 357–406. [Google Scholar]
- Nikonowicz, EP. Preparation and use of 2H-labeled RNA oligonucleotides in nuclear magnetic resonance studies. Methods Enzymol 2001, 338, 320–341. [Google Scholar]
- Scott, LG; Tolbert, TJ; Williamson, JR. Preparation of specifically 2H- and 13C-labeled ribonucleotides. Methods Enzymol 2000, 317, 18–38. [Google Scholar]
- Davis, JH; Tonelli, M; Scott, LG; Jaeger, L; Williamson, JR; Butcher, SE. RNA helical packing in solution: NMR structure of a 30 kDa GAAA tetraloop-receptor complex. J Mol Biol 2005, 351, 371–382, Erratum in: J. Mol. Biol. 2006, 360, 742. [Google Scholar]
- Tolbert, TJ; Williamson, JR. Preparation of Specifically Deuterated RNA for NMR Studies Using a Combination of Chemical and Enzymatic Synthesis. J Am Chem Soc 1996, 118, 7929–7940. [Google Scholar]
- Vallurupalli, P; Scott, L; Hennig, M; Williamson, JR; Kay, LE. New RNA labeling methods offer dramatic sensitivity enhancements in 2H NMR relaxation spectra. J Am Chem Soc 2006, 128, 9346–9347. [Google Scholar]
- Hirschbein, BL; Mazenod, FP; Whitesides, GM. Synthesis of phosphoenolypyruvate and its use in ATP cofactor regeneration. J Org Chem 1982, 47, 3765–3766. [Google Scholar]
- Simon, ES; Grabowski, S; Whitesides, GM. Preparation of phosphoenolpyruvate from D-(−)-3-phosphoglyceric acid for use in regeneration of ATP. J Am Chem Soc 1989, 111, 8920–8921. [Google Scholar]
- Rising, KA; Schramm, VL. Enzymic Synthesis of NAD+ with the Specific Incorporation of Atomic Labels. J Am Chem Soc 1994, 116, 6531–6536. [Google Scholar]
- Scott, LG; Geierstanger, BH; Williamson, JR; Hennig, M. Enzymatic synthesis and 19F NMR studies of 2-fluoroadenine-substituted RNA. J Am Chem Soc 2004, 126, 11776–11777. [Google Scholar]
- Hennig, M; Scott, LG; Sperling, E; Bermel, W; Williamson, JR. Synthesis of 5-fluoropyrimidine nucleotides as sensitive NMR probes of RNA structure. J Am Chem Soc 2007, 129, 14911–14921. [Google Scholar]
- Toth, EA. Molecular replacement. Methods Mol Biol 2007, 364, 121–148. [Google Scholar]
- Golden, BL; Gooding, AR; Podell, ER; Cech, TR. X-ray crystallography of large RNAs: heavy-atom derivatives by RNA engineering. RNA 1996, 2, 1295–1305. [Google Scholar]
- Golden, BL. Heavy atom derivatives of RNA. Methods Enzymol 2000, 317, 124–132. [Google Scholar]
- Wedekind, JE; McKay, DB. Purification, crystallization, and X-ray diffraction analysis of small ribozymes. Methods Enzymol 2000, 317, 149–168. [Google Scholar]
- Correll, CC; Freeborn, B; Moore, PB; Steitz, TA. Use of chemically modified nucleotides to determine a 62-nucleotide RNA crystal structure: a survey of phosphorothioates, Br, Pt and Hg. J Biomol Struct Dyn 1997, 15, 165–172. [Google Scholar]
- Baugh, C; Grate, D; Wilson, C. 2.8 A crystal structure of the malachite green aptamer. J Mol Biol 2000, 301, 117–128. [Google Scholar]
- Martick, M; Scott, WG. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell 2006, 126, 309–320. [Google Scholar]
- Ferre-D'Amare, AR; Doudna, JA. Crystallization and structure determination of a hepatitis delta virus ribozyme: use of the RNA-binding protein U1A as a crystallization module. J Mol Biol 2000, 295, 541–556. [Google Scholar]
- Ferre-D'Amare, AR; Zhou, K; Doudna, JA. Crystal structure of a hepatitis delta virus ribozyme. Nature 1998, 395, 567–574. [Google Scholar]
- Sheng, J; Huang, Z. Selenium Derivatization of Nucleic Acids for Phase and Structure Determination in Nucleic Acid X-ray Crystallography. Int J Mol Sci 2008, 9, 258–271. [Google Scholar]
- Brandt, G; Carrasco, N; Huang, Z. Efficient substrate cleavage catalyzed by hammerhead ribozymes derivatized with selenium for X-ray crystallography. Biochemistry 2006, 45, 8972–8977. [Google Scholar]
- Keel, AY; Rambo, RP; Batey, RT; Kieft, JS. A general strategy to solve the phase problem in RNA crystallography. Structure 2007, 15, 761–772. [Google Scholar]
- Sokolova, NI; Ashirbekova, DT; Dolinnaya, NG; Shabarova, ZA. Chemical reactions within DNA duplexes. Cyanogen bromide as an effective oligodeoxyribonucleotide coupling agent. FEBS Lett 1988, 232, 153–155. [Google Scholar]
- Dolinnaya, NG; Sokolova, NI; Ashirbekova, DT; Shabarova, ZA. The use of BrCN for assembling modified DNA duplexes and DNA-RNA hybrids; comparison with water-soluble carbodiimide. Nucleic Acids Res 1991, 19, 3067–3072. [Google Scholar]
- Mitra, D; Damha, MJ. A novel approach to the synthesis of DNA and RNA lariats. J Org Chem 2007, 72, 9491–9500. [Google Scholar]
- Moore, MJ; Query, CC. Joining of RNAs by splinted ligation. Methods Enzymol 2000, 317, 109–123. [Google Scholar]
- Romaniuk, PJ; Uhlenbeck, OC. Joining of RNA molecules with RNA ligase. Methods Enzymol 1983, 100, 52–59. [Google Scholar]
- Tzakos, AG; Easton, LE; Lukavsky, PJ. Preparation of large RNA oligonucleotides with complementary isotope-labeled segments for NMR structural studies. Nat Protoc 2007, 2, 2139–2147. [Google Scholar]
- Stark, MR; Pleiss, JA; Deras, M; Scaringe, SA; Rader, SD. An RNA ligase-mediated method for the efficient creation of large, synthetic RNAs. RNA 2006, 12, 2014–2019. [Google Scholar]
- Kim, I; Lukavsky, PJ; Puglisi, JD. NMR study of 100 kDa HCV IRES RNA using segmental isotope labeling. J Am Chem Soc 2002, 124, 9338–9339. [Google Scholar]
- Bain, JD; Switzer, C. Regioselective ligation of oligoribonucleotides using DNA splints. Nucleic Acids Res 1992, 20, 4372. [Google Scholar]
- Höbartner, C; Silverman, SK. Recent advances in DNA catalysis. Biopolymers 2007, 87, 279–292. [Google Scholar]
- Purtha, WE; Coppins, RL; Smalley, MK; Silverman, SK. General deoxyribozyme-catalyzed synthesis of native 3′-5′ RNA linkages. J Am Chem Soc 2005, 127, 13124–13125. [Google Scholar]
- D'Souza, V; Summers, MF. How retroviruses select their genomes. Nat Rev Microbiol 2005, 3, 643–655. [Google Scholar]
- D'Souza, V; Summers, MF. Structural basis for packaging the dimeric genome of Moloney murine leukaemia virus. Nature 2004, 431, 586–590. [Google Scholar]
- Pyle, AM; Lambowitz, AM. Group II introns: Ribozymes that splice RNA and invade DNA. In RNA WorldGesteland, RF, Cech, TR, Atkins, JF, Eds.; Cold Spring Harbor Laboratory Press: New York, 2006, 3rd Ed; Chapter 17; p. 469. [Google Scholar]
- Gumbs, OH; Padgett, RA; Dayie, KT. Fluorescence and solution NMR study of the active site of a 160-kDa group II intron ribozyme. RNA 2006, 12, 1693–1707. [Google Scholar]
- Seetharaman, M; Eldho, NV; Padgett, RA; Dayie, KT. Structure of a self-splicing group II intron catalytic effector domain 5: parallels with spliceosomal U6 RNA. RNA 2006, 12, 235–247. [Google Scholar]
- Eldho, NV; Dayie, KT. Internal bulge and tetraloop of the catalytic domain 5 of a group II intron ribozyme are flexible: implications for catalysis. J Mol Biol 2007, 365, 930–944. [Google Scholar]
- Dayie, KT; Gumbs, OH; Eldho, NV; Seetharaman, M; Thompson, M. In-gel fluorescence probing of RNA-RNA interactions. Anal Biochem 2007, 362, 278–280. [Google Scholar]
- Ying, BW; Fourmy, D; Yoshizawa, S. Substitution of the use of radioactivity by fluorescence for biochemical studies of RNA. RNA 2007, 13, 2042–2050. [Google Scholar]
- Szymczyna, BR; Gan, L; Johnson, JE; Williamson, JR. Solution NMR studies of the maturation intermediates of a 13 MDa viral capsid. J Am Chem Soc 2007, 129, 7867–7876. [Google Scholar]
- Toor, N; Keating, KS; Taylor, SD; Pyle, AM. Crystal structure of a self-spliced group II intron. Science 2008, 320, 77–82. [Google Scholar]
- Dayie, KT; Padgett, RA. A glimpse into the active site of the Group II intron and maybe the spliceosome too. In RNA; 2008; in press. [Google Scholar]
Share and Cite
Dayie, K.T. Key Labeling Technologies to Tackle Sizeable Problems in RNA Structural Biology. Int. J. Mol. Sci. 2008, 9, 1214-1240. https://doi.org/10.3390/ijms9071214
Dayie KT. Key Labeling Technologies to Tackle Sizeable Problems in RNA Structural Biology. International Journal of Molecular Sciences. 2008; 9(7):1214-1240. https://doi.org/10.3390/ijms9071214
Chicago/Turabian StyleDayie, Kwaku T. 2008. "Key Labeling Technologies to Tackle Sizeable Problems in RNA Structural Biology" International Journal of Molecular Sciences 9, no. 7: 1214-1240. https://doi.org/10.3390/ijms9071214
APA StyleDayie, K. T. (2008). Key Labeling Technologies to Tackle Sizeable Problems in RNA Structural Biology. International Journal of Molecular Sciences, 9(7), 1214-1240. https://doi.org/10.3390/ijms9071214