The Molecular Structure and Vibrational Spectrum of 6-bromo-8-thia-1,4-epoxybicyclo[4.3.0]non-2-ene

Abstract
:1. Introduction
2. Experimental
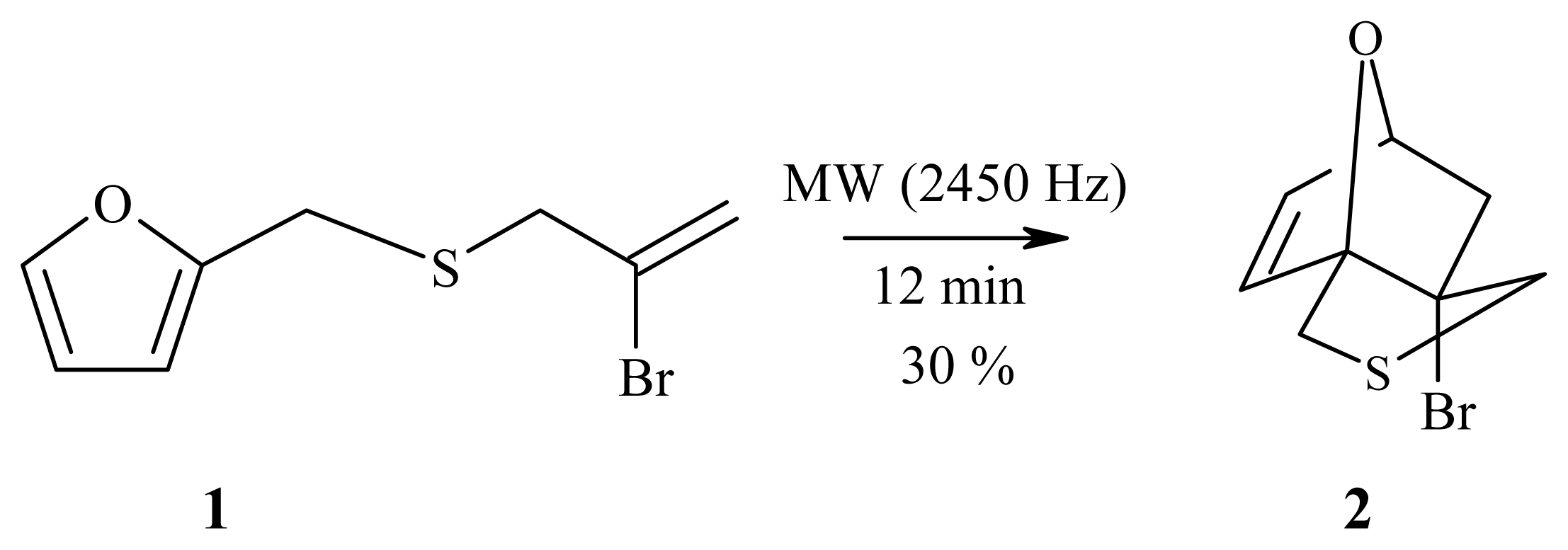
2.1. Synthesis of 6-bromo-8-thia-1,4-epoxybicyclo[4.3.0]non-2-ene
2.2. Instrumentation
2.2. Calculations details
3. Results and Discussion
3.1. Geometry optimization
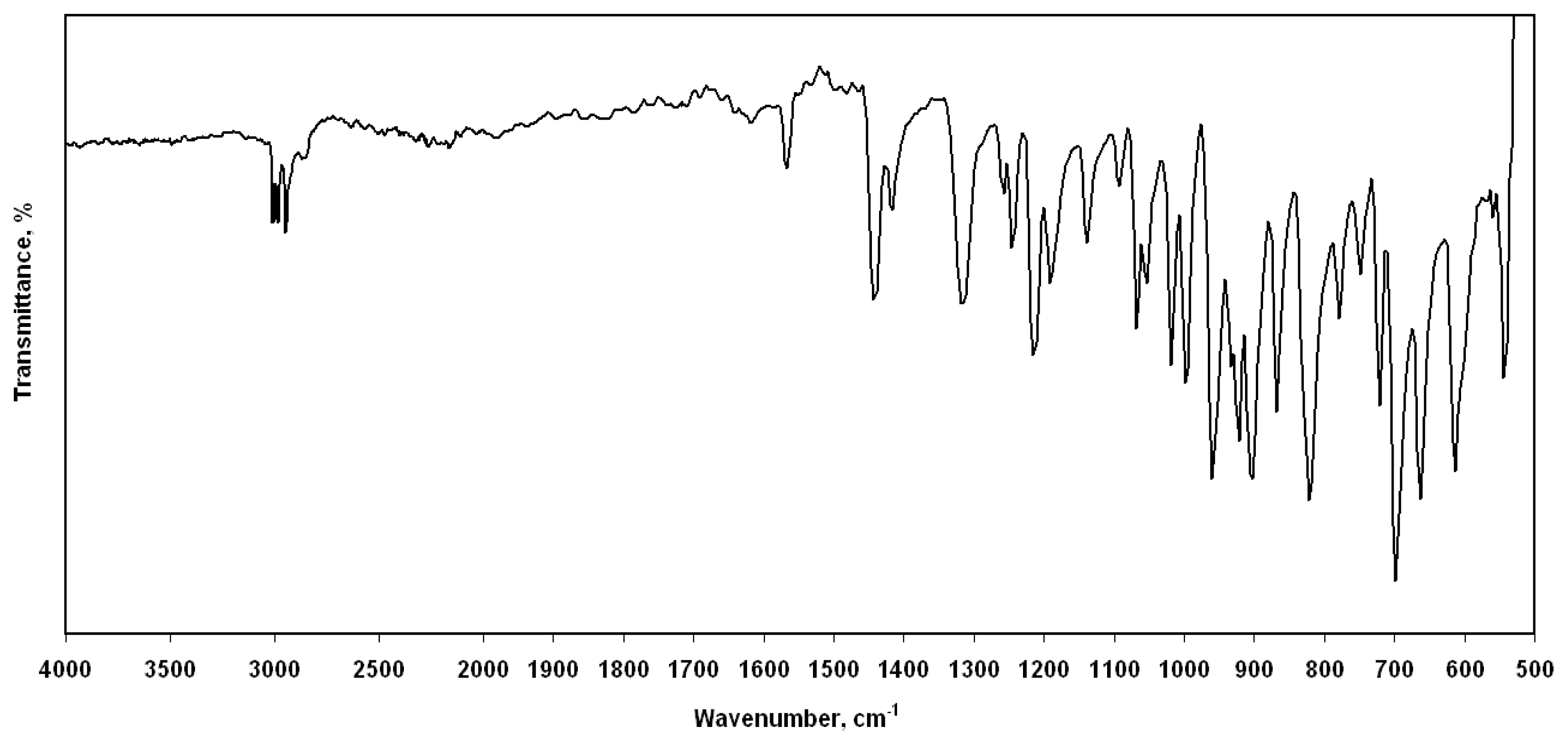
3.2. Vibrational frequencies
4. Conclusions


{kind=link}
{kind=link}
{kind=link}
| Parameters | Experimental | Calculated | |||
|---|---|---|---|---|---|
| HF | DFT | ||||
| B3LYP | B3PW91 | mPW1PW91 | |||
| Bond lengths (Å) | |||||
| R(1,4) | 1.508(6) | 1.529 | 1.529 | 1.524 | 1.522 |
| R(1,19) | 1.813(4) | 1.824 | 1.844 | 1.829 | 1.825 |
| R(4,5) | 1.533(5) | 1.548 | 1.551 | 1.544 | 1.541 |
| R(4,14) | 1.552(5) | 1.557 | 1.575 | 1.569 | 1.566 |
| R(4,20) | 1.965(3) | 1.964 | 1.989 | 1.967 | 1.959 |
| R(5,8) | 1.554(6) | 1.556 | 1.570 | 1.564 | 1.561 |
| R(8,10) | 1.512(8) | 1.520 | 1.522 | 1.517 | 1.515 |
| R(8,18) | 1.436(5) | 1.415 | 1.443 | 1.435 | 1.431 |
| R(10,12) | 1.314(7) | 1.317 | 1.335 | 1.334 | 1.332 |
| R(12,14) | 1.508(6) | 1.521 | 1.524 | 1.520 | 1.517 |
| R(14,15) | 1.502(5) | 1.513 | 1.515 | 1.512 | 1.510 |
| R(14,18) | 1.444(4) | 1.414 | 1.443 | 1.435 | 1.430 |
| R(15,19) | 1.829(4) | 1.829 | 1.852 | 1.839 | 1.834 |
| r | 0.9971 | 0.9994 | 0.9990 | 0.9988 | |
| Bond angles (°) | |||||
| A(4,1,19) | 107.7(3) | 107.53 | 107.69 | 107.49 | 107.44 |
| A(1,4,5) | 115.5(3) | 115.02 | 115.45 | 115.30 | 115.15 |
| A(1,4,14) | 106.5(3) | 105.48 | 105.63 | 105.26 | 105.16 |
| A(1,4,20) | 109.1(3) | 109.46 | 109.41 | 109.62 | 109.72 |
| A(5,4,14) | 102.3(3) | 101.62 | 101.98 | 101.95 | 101.93 |
| A(5,4,20) | 113.0(3) | 113.10 | 112.78 | 113.15 | 113.31 |
| A(14,4,20) | 109.9(2) | 111.68 | 111.12 | 111.01 | 110.97 |
| A(4,5,8) | 100.0(3) | 99.59 | 99.87 | 99.77 | 99.73 |
| A(5,8,10) | 107.1(4) | 107.56 | 107.32 | 106.98 | 106.87 |
| A(5,8,18) | 100.9(3) | 101.13 | 101.13 | 101.34 | 101.41 |
| A(10,8,18) | 102.0(4) | 101.42 | 101.89 | 102.11 | 102.10 |
| A(8,10,12) | 105.9(4) | 105.45 | 105.53 | 105.39 | 105.37 |
| A(10,12,14) | 105.7(4) | 105.38 | 105.55 | 105.33 | 105.28 |
| A(4,14,12) | 109.9(3) | 110.24 | 109.59 | 109.24 | 109.22 |
| A(4,14,15) | 111.0(3) | 110.96 | 111.01 | 110.77 | 110.72 |
| A(4,14,18) | 97.3(3) | 97.64 | 97.51 | 97.73 | 97.79 |
| A(12,14,15) | 121.4(3) | 120.76 | 121.16 | 121.23 | 121.20 |
| A(12,14,18) | 102.1(3) | 101.60 | 102.02 | 102.27 | 102.30 |
| A(15,14,18) | 112.3(3) | 112.88 | 112.69 | 112.81 | 112.86 |
| A(14,15,19) | 107.4(3) | 107.52 | 107.97 | 107.99 | 107.94 |
| A(8,18,14) | 95.7(3) | 97.39 | 96.37 | 96.38 | 96.44 |
| A(1,19,15) | 94.58(18) | 94.50 | 93.75 | 93.81 | 93.86 |
| r | 0.9943 | 0.9975 | 0.9965 | 0.9961 | |
| No | Wave number | IR intensity | Red mass | Force Const. | Assignments, PED (%)d | ||||
|---|---|---|---|---|---|---|---|---|---|
| Exp. | Unscal. | Scalb | Scalc | Abs. | Rel. | ||||
| 1 | 3140 | 3261 | 3115 | 3135 | 4 | 12 | 1.11 | 6.93 | νCH, C10,12, sym (98) |
| 2 | 3077 | 3233 | 3088 | 3109 | 2 | 8 | 1.09 | 6.71 | νCH, C10,12, asym (100) |
| 3 | 3008 | 3156 | 3015 | 3035 | 3 | 11 | 1.11 | 6.49 | νCH, C1, asym (88) |
| 4 | 3008 | 3153 | 3011 | 3031 | 8 | 26 | 1.10 | 6.46 | νCH, C5, asym (82) |
| 5 | 2983 | 3149 | 3007 | 3027 | 1 | 5 | 1.11 | 6.48 | νCH, C15, asym (100) |
| 6 | 2983 | 3148 | 3007 | 3026 | 30 | 99 | 1.09 | 6.37 | νCH, C8, (92) |
| 7 | 2948 | 3093 | 2954 | 2973 | 12 | 40 | 1.06 | 5.96 | νCH, C15, sym (100) |
| 8 | 2948 | 3091 | 2952 | 2971 | 29 | 95 | 1.06 | 5.98 | νCH, C1,5, sym (90) |
| 9 | 2933 | 3084 | 2946 | 2965 | 1 | 4 | 1.06 | 5.95 | νCH, C1,5, sym (93) |
| 10 | 1568 | 1657 | 1583 | 1593 | 2 | 8 | 6.40 | 10.36 | νC=C (83) |
| 11 | 1447 | 1503 | 1435 | 1445 | 5 | 15 | 1.09 | 1.45 | δCH2, scis, C5 (92) |
| 12 | 1439 | 1486 | 1420 | 1429 | 11 | 37 | 1.10 | 1.44 | δCH2, scis, C15 (74) |
| 13 | 1418 | 1476 | 1410 | 1419 | 5 | 16 | 1.09 | 1.40 | δCH2, scis, C1 (78) |
| 14 | 1318 | 1352 | 1292 | 1300 | 12 | 40 | 1.97 | 2.13 | δ=CH, ipb (64) |
| 15 | 1310 | 1346 | 1285 | 1294 | 6 | 20 | 2.04 | 2.18 | δOCH(22)+δCH, ipb (11) + νCC, C14,15(13) |
| 16 | 1259 | 1320 | 1260 | 1269 | 0 | 1 | 1.49 | 1.53 | δOCH (40) + δCH2, wagg, C5 (34) |
| 17 | 1246 | 1294 | 1236 | 1244 | 6 | 19 | 1.34 | 1.33 | δCH2, wagg, C1,15 (59) |
| 18 | 1218 | 1279 | 1222 | 1230 | 14 | 47 | 1.46 | 1.40 | δ=CH, ipb (12) + δCH2, wagg, C1,15 (27) |
| 19 | 1211 | 1250 | 1194 | 1202 | 18 | 59 | 1.49 | 1.37 | δCH2, wagg, C5 (42) + δOCH (12) |
| 20 | 1193 | 1237 | 1182 | 1190 | 5 | 17 | 1.67 | 1.51 | νCC, C14,15 (14) + δCH2, wagg, C15 (20) |
| 21 | 1189 | 1213 | 1158 | 1166 | 13 | 44 | 1.57 | 1.36 | δCH2, twist, C1,5 (36) + νCC, C1,4 (14) |
| 22 | 1140 | 1207 | 1152 | 1160 | 3 | 9 | 1.39 | 1.19 | δCH2, twist, C15 (45) |
| 23 | 1094 | 1162 | 1110 | 1118 | 5 | 15 | 1.23 | 0.98 | δCH2, twist, C1 (34) |
| 24 | 1068 | 1114 | 1064 | 1071 | 2 | 8 | 1.67 | 1.22 | νCC (25) + δCH (37) |
| 25 | 1055 | 1094 | 1045 | 1052 | 9 | 30 | 1.48 | 1.04 | δ=CH, ipb (47) |
| 26 | 1019 | 1071 | 1023 | 1030 | 12 | 38 | 2.61 | 1.76 | νCC, (21) + δCCC (12) |
| 27 | 998 | 1037 | 991 | 997 | 19 | 62 | 2.96 | 1.88 | νCC, C4,5 (28) + δ=CH, ipb (28) |
| 28 | 962 | 1021 | 975 | 981 | 19 | 61 | 2.25 | 1.38 | δCH2, rock, C1,5,15 (24) |
| 29 | 934 | 981 | 937 | 943 | 30 | 100 | 2.30 | 1.30 | δCH2, rock, C15 (23) + δCOC (13) |
| 30 | 923 | 952 | 910 | 915 | 20 | 67 | 2.96 | 1.58 | δCH2, rock, C1 (34) |
| 31 | 909 | 938 | 895 | 901 | 5 | 17 | 1.53 | 0.79 | δCH2, opb (71) |
| 32 | 903 | 928 | 886 | 892 | 22 | 73 | 2.11 | 1.07 | νCC, (47) + δ=CC, opb (33) |
| 33 | 870 | 912 | 871 | 877 | 11 | 36 | 2.09 | 1.02 | νCC, (19) + δCH2, rock, C1,5 (42) |
| 34 | 830 | 880 | 841 | 846 | 6 | 21 | 2.37 | 1.08 | νCC(16) + δCOC,(20) + δCH2,rock, C1,5 (10) |
| 35 | 822 | 840 | 803 | 808 | 3 | 11 | 2.58 | 1.07 | νCC(10) + δCOC,(27) + δCH2,rock, C15 (41) |
| 36 | 780 | 836 | 799 | 804 | 15 | 50 | 3.57 | 1.47 | νCO (25) + δ=CC, (33) |
| 37 | 750 | 786 | 751 | 756 | 3 | 11 | 3.55 | 1.29 | δCCC, (26) + νCC (10) |
| 38 | 722 | 752 | 718 | 723 | 5 | 15 | 4.21 | 1.40 | νCS (28) + γC, C14, (15) |
| 39 | 701 | 730 | 698 | 702 | 3 | 11 | 2.74 | 0.86 | νCS (10) + δCCS, (10) + γCH, C10,12 (26) |
| 40 | 688 | 723 | 690 | 695 | 9 | 29 | 4.80 | 1.48 | νCS (14) + δCCC, (24) + γCH, C14 (28) |
| 41 | 666 | 709 | 677 | 681 | 30 | 97 | 2.21 | 0.65 | γCH, C10,12 (35) |
| 42 | 650 | 672 | 641 | 646 | 5 | 17 | 4.11 | 1.09 | νCC (17) + γCH (13) + δCCO, (16) |
| 43 | 617 | 621 | 593 | 597 | 13 | 43 | 4.77 | 1.08 | δOCC, (21) + τCO, (14) |
| 44 | 543 | 544 | 520 | 523 | 6 | 21 | 2.96 | 0.52 | τCH (39) |
| 45 | - | 505 | 482 | 485 | 3 | 9 | 4.79 | 0.72 | νCC (11) + νCS (14) + δSCC, (16) |
| 46 | - | 402 | 384 | 387 | 2 | 8 | 2.57 | 0.25 | δCCO, (11) + τCO, (13) |
| 47 | - | 387 | 370 | 372 | 1 | 3 | 5.34 | 0.47 | δSCC, (13) + νCS (10) + νCO (13) |
| 48 | - | 345 | 329 | 331 | 1 | 4 | 4.19 | 0.29 | τCC, (27) + γC (10) |
| 49 | - | 325 | 311 | 313 | 1 | 5 | 3.55 | 0.22 | νCBr (44) + δCCO, (18) |
| 50 | - | 282 | 269 | 271 | 1 | 4 | 2.44 | 0.11 | γC, C4 (43) + δCCC, (17) |
| 51 | - | 262 | 250 | 252 | 2 | 8 | 3.93 | 0.16 | νCBr (27) + δCCC, (12) |
| 52 | - | 144 | 138 | 139 | 2 | 6 | 5.71 | 0.07 | δCCBr, (25) + γC, C4 (36) |
| 53 | - | 116 | 110 | 111 | 1 | 3 | 5.97 | 0.05 | δCCBr, (28) + τCC (43) |
| 54 | - | 78 | 75 | 75 | 2 | 6 | 5.11 | 0.02 | τCC (52) |
| r | 0.9999 | 0.9999 | 0.9999 | ||||||
| Mean dev. | 63.71 | −2.83 | −57.54 | ||||||
| Mean abs. deviation | 63.71 | 12.22 | 57.54 | ||||||
| Ave. absolute error | 4.27 | 1.09 | 4.25 | ||||||
| RMSmol | 78.8 | 14.6 | 65.3 | ||||||
| RMSover | 71.1 | 13.2 | 58.9 | ||||||
| Sca. Factor | 1.0000 | 0.9552 | 0.9614 | ||||||
| Number | Wave number | IR intensity | Red Mass | Force Constant | ||||
|---|---|---|---|---|---|---|---|---|
| Exp. | Unscaled | Scaledb | Scaledc | Abs. | Rel. | |||
| 1 | 3140 | 3423 | 3064 | 3078 | 4 | 11 | 1.11 | 7.66 |
| 2 | 3077 | 3393 | 3037 | 3051 | 3 | 7 | 1.09 | 7.39 |
| 3 | 3008 | 3319 | 2971 | 2984 | 4 | 9 | 1.11 | 7.20 |
| 4 | 3008 | 3312 | 2964 | 2978 | 42 | 100 | 1.09 | 7.06 |
| 5 | 2983 | 3309 | 2962 | 2976 | 4 | 10 | 1.11 | 7.16 |
| 6 | 2983 | 3307 | 2960 | 2973 | 9 | 22 | 1.10 | 7.10 |
| 7 | 2948 | 3257 | 2916 | 2929 | 25 | 59 | 1.06 | 6.62 |
| 8 | 2948 | 3251 | 2910 | 2923 | 22 | 53 | 1.06 | 6.60 |
| 9 | 2933 | 3239 | 2900 | 2913 | 12 | 28 | 1.07 | 6.59 |
| 10 | 1568 | 1816 | 1625 | 1633 | 3 | 7 | 6.28 | 12.19 |
| 11 | 1447 | 1630 | 1459 | 1466 | 4 | 9 | 1.09 | 1.71 |
| 12 | 1439 | 1615 | 1445 | 1452 | 12 | 30 | 1.11 | 1.71 |
| 13 | 1418 | 1610 | 1442 | 1448 | 2 | 6 | 1.10 | 1.68 |
| 14 | 1318 | 1486 | 1330 | 1336 | 18 | 43 | 2.24 | 2.91 |
| 15 | 1310 | 1481 | 1326 | 1332 | 7 | 16 | 1.83 | 2.36 |
| 16 | 1259 | 1458 | 1305 | 1311 | 1 | 2 | 1.55 | 1.94 |
| 17 | 1246 | 1432 | 1282 | 1287 | 11 | 26 | 1.39 | 1.68 |
| 18 | 1218 | 1409 | 1261 | 1267 | 12 | 29 | 1.46 | 1.71 |
| 19 | 1211 | 1392 | 1246 | 1252 | 23 | 55 | 1.56 | 1.78 |
| 20 | 1193 | 1355 | 1213 | 1219 | 5 | 13 | 1.93 | 2.09 |
| 21 | 1189 | 1331 | 1192 | 1197 | 4 | 11 | 1.42 | 1.48 |
| 22 | 1140 | 1327 | 1188 | 1193 | 22 | 54 | 1.62 | 1.67 |
| 23 | 1094 | 1274 | 1141 | 1146 | 7 | 16 | 1.24 | 1.19 |
| 24 | 1068 | 1214 | 1087 | 1092 | 5 | 13 | 1.66 | 1.44 |
| 25 | 1055 | 1205 | 1079 | 1084 | 26 | 62 | 2.27 | 1.94 |
| 26 | 1019 | 1182 | 1058 | 1063 | 9 | 22 | 2.52 | 2.08 |
| 27 | 998 | 1160 | 1039 | 1043 | 15 | 37 | 1.83 | 1.45 |
| 28 | 962 | 1130 | 1012 | 1016 | 38 | 91 | 2.46 | 1.85 |
| 29 | 934 | 1103 | 987 | 991 | 28 | 68 | 2.05 | 1.47 |
| 30 | 923 | 1070 | 958 | 963 | 2 | 5 | 1.39 | 0.94 |
| 31 | 909 | 1045 | 935 | 939 | 27 | 66 | 2.54 | 1.63 |
| 32 | 903 | 1023 | 916 | 920 | 20 | 47 | 2.18 | 1.34 |
| 33 | 870 | 1000 | 895 | 899 | 14 | 33 | 2.01 | 1.18 |
| 34 | 830 | 955 | 855 | 859 | 5 | 11 | 2.51 | 1.35 |
| 35 | 822 | 932 | 835 | 838 | 17 | 41 | 2.19 | 1.12 |
| 36 | 780 | 914 | 818 | 822 | 2 | 4 | 3.10 | 1.53 |
| 37 | 750 | 871 | 780 | 783 | 7 | 17 | 3.51 | 1.57 |
| 38 | 722 | 837 | 749 | 753 | 3 | 8 | 4.72 | 1.95 |
| 39 | 701 | 815 | 729 | 733 | 27 | 65 | 1.69 | 0.66 |
| 40 | 688 | 801 | 717 | 720 | 10 | 25 | 4.43 | 1.67 |
| 41 | 666 | 794 | 711 | 714 | 26 | 62 | 3.75 | 1.39 |
| 42 | 650 | 728 | 652 | 655 | 4 | 10 | 4.25 | 1.33 |
| 43 | 617 | 681 | 609 | 612 | 14 | 33 | 4.77 | 1.30 |
| 44 | 543 | 593 | 531 | 533 | 8 | 19 | 2.89 | 0.60 |
| 45 | - | 548 | 491 | 493 | 3 | 6 | 4.92 | 0.87 |
| 46 | - | 440 | 394 | 396 | 2 | 5 | 2.61 | 0.30 |
| 47 | - | 418 | 375 | 376 | 1 | 3 | 5.31 | 0.55 |
| 48 | - | 373 | 334 | 335 | 2 | 4 | 4.06 | 0.33 |
| 49 | - | 355 | 318 | 319 | 2 | 5 | 3.53 | 0.26 |
| 50 | - | 307 | 275 | 276 | 1 | 3 | 2.51 | 0.14 |
| 51 | - | 288 | 258 | 259 | 3 | 7 | 3.92 | 0.19 |
| 52 | - | 160 | 143 | 144 | 2 | 5 | 5.46 | 0.08 |
| 53 | - | 128 | 114 | 115 | 1 | 3 | 5.38 | 0.05 |
| 54 | - | 89 | 80 | 80 | 2 | 4 | 6.01 | 0.03 |
| r | 0.9997 | 0.9997 | 0.9997 | |||||
| Mean deviation | 181.57 | 13.64 | −130.89 | |||||
| Mean absolute deviation | 181.57 | 30.20 | 130.89 | |||||
| Average absolute error | 13.83 | 2.57 | 8.38 | |||||
| RMSmol | 196.0 | 34.0 | 169.6 | |||||
| RMSover | 177.0 | 30.7 | 153.1 | |||||
| Scaling Factor | 1.0000 | 0.8952 | 0.8992 | |||||
| Number | Wave number | IR intensity | Red mass | Force Constant | ||||
|---|---|---|---|---|---|---|---|---|
| Exp. | Unscaled | Scaledb | Scaledc | Abs. | Rel. | |||
| 1 | 3140 | 3274 | 3117 | 3134 | 3 | 8 | 1.11 | 6.99 |
| 2 | 3077 | 3246 | 3090 | 3107 | 2 | 6 | 1.09 | 6.76 |
| 3 | 3008 | 3167 | 3015 | 3031 | 6 | 17 | 1.11 | 6.53 |
| 4 | 3008 | 3164 | 3013 | 3029 | 2 | 7 | 1.11 | 6.52 |
| 5 | 2983 | 3161 | 3010 | 3026 | 1 | 3 | 1.11 | 6.53 |
| 6 | 2983 | 3159 | 3007 | 3024 | 29 | 83 | 1.09 | 6.41 |
| 7 | 2948 | 3101 | 2952 | 2969 | 13 | 36 | 1.06 | 6.00 |
| 8 | 2948 | 3099 | 2950 | 2966 | 21 | 61 | 1.06 | 6.01 |
| 9 | 2933 | 3091 | 2943 | 2959 | 5 | 15 | 1.06 | 5.97 |
| 10 | 1568 | 1666 | 1586 | 1595 | 3 | 7 | 6.53 | 10.67 |
| 11 | 1447 | 1493 | 1421 | 1429 | 6 | 17 | 1.09 | 1.43 |
| 12 | 1439 | 1478 | 1407 | 1415 | 13 | 38 | 1.11 | 1.43 |
| 13 | 1418 | 1466 | 1395 | 1403 | 7 | 19 | 1.09 | 1.38 |
| 14 | 1318 | 1356 | 1291 | 1299 | 16 | 47 | 2.53 | 2.75 |
| 15 | 1310 | 1350 | 1285 | 1292 | 4 | 12 | 1.87 | 2.01 |
| 16 | 1259 | 1320 | 1257 | 1264 | 0 | 0 | 1.57 | 1.61 |
| 17 | 1246 | 1293 | 1231 | 1238 | 4 | 12 | 1.39 | 1.37 |
| 18 | 1218 | 1277 | 1216 | 1223 | 12 | 34 | 1.46 | 1.41 |
| 19 | 1211 | 1252 | 1192 | 1198 | 19 | 55 | 1.65 | 1.52 |
| 20 | 1193 | 1239 | 1179 | 1186 | 4 | 12 | 1.54 | 1.39 |
| 21 | 1189 | 1216 | 1158 | 1164 | 15 | 43 | 1.64 | 1.43 |
| 22 | 1140 | 1209 | 1151 | 1157 | 3 | 9 | 1.41 | 1.21 |
| 23 | 1094 | 1161 | 1105 | 1111 | 5 | 15 | 1.24 | 0.98 |
| 24 | 1068 | 1118 | 1065 | 1071 | 1 | 3 | 1.69 | 1.24 |
| 25 | 1055 | 1098 | 1045 | 1051 | 16 | 44 | 1.61 | 1.14 |
| 26 | 1019 | 1080 | 1028 | 1034 | 9 | 25 | 2.27 | 1.56 |
| 27 | 998 | 1047 | 997 | 1002 | 17 | 49 | 2.57 | 1.66 |
| 28 | 962 | 1030 | 981 | 986 | 24 | 70 | 2.34 | 1.47 |
| 29 | 934 | 993 | 945 | 950 | 27 | 78 | 2.31 | 1.34 |
| 30 | 923 | 961 | 915 | 920 | 20 | 57 | 2.80 | 1.52 |
| 31 | 909 | 943 | 898 | 903 | 17 | 49 | 1.93 | 1.01 |
| 32 | 903 | 934 | 889 | 894 | 11 | 33 | 1.57 | 0.81 |
| 33 | 870 | 919 | 875 | 880 | 12 | 35 | 2.15 | 1.07 |
| 34 | 830 | 886 | 843 | 848 | 6 | 16 | 2.17 | 1.00 |
| 35 | 822 | 847 | 807 | 811 | 12 | 35 | 2.62 | 1.11 |
| 36 | 780 | 845 | 805 | 809 | 1 | 2 | 3.13 | 1.32 |
| 37 | 750 | 794 | 756 | 760 | 2 | 7 | 3.42 | 1.27 |
| 38 | 722 | 767 | 730 | 734 | 4 | 11 | 4.73 | 1.64 |
| 39 | 701 | 743 | 707 | 711 | 8 | 23 | 4.87 | 1.58 |
| 40 | 688 | 737 | 701 | 705 | 3 | 10 | 2.85 | 0.91 |
| 41 | 666 | 715 | 681 | 684 | 35 | 100 | 1.98 | 0.60 |
| 42 | 650 | 676 | 644 | 647 | 6 | 17 | 4.05 | 1.09 |
| 43 | 617 | 625 | 595 | 598 | 10 | 30 | 4.60 | 1.06 |
| 44 | 543 | 546 | 520 | 522 | 6 | 17 | 2.93 | 0.51 |
| 45 | - | 508 | 484 | 487 | 3 | 7 | 4.81 | 0.73 |
| 46 | - | 402 | 382 | 385 | 3 | 7 | 2.67 | 0.25 |
| 47 | - | 389 | 370 | 372 | 1 | 3 | 5.23 | 0.47 |
| 48 | - | 348 | 331 | 333 | 2 | 5 | 4.25 | 0.30 |
| 49 | - | 329 | 313 | 315 | 1 | 3 | 3.37 | 0.21 |
| 50 | - | 282 | 268 | 270 | 1 | 3 | 2.40 | 0.11 |
| 51 | - | 264 | 251 | 253 | 2 | 6 | 3.79 | 0.16 |
| 52 | - | 144 | 137 | 138 | 2 | 5 | 5.69 | 0.07 |
| 53 | - | 115 | 110 | 110 | 1 | 2 | 6.47 | 0.05 |
| 54 | - | 76 | 72 | 73 | 2 | 6 | 4.75 | 0.02 |
| r | 0.9998 | 0.9998 | 0.9998 | |||||
| Mean deviation | 69.36 | −2.08 | −62.63 | |||||
| Mean absolute deviation | 69.36 | 13.98 | 62.63 | |||||
| Average absolute error | 4.79 | 1.26 | 4.50 | |||||
| RMSmol | 84.6 | 16.4 | 72.3 | |||||
| RMSover | 76.4 | 14.8 | 65.2 | |||||
| Scaling Factor | 1.0000 | 0.9520 | 0.9573 | |||||
| Number | Wave number | IR intensity | Red mass | Force Constant | ||||
|---|---|---|---|---|---|---|---|---|
| Exp. | Unscaled | Scaledb | Scaledc | Abs. | Rel. | |||
| 1 | 3140 | 3293 | 3114 | 3129 | 2 | 6 | 1.11 | 7.08 |
| 2 | 3077 | 3266 | 3088 | 3102 | 2 | 4 | 1.09 | 6.84 |
| 3 | 3008 | 3185 | 3012 | 3026 | 6 | 17 | 1.11 | 6.61 |
| 4 | 3008 | 3183 | 3010 | 3024 | 2 | 6 | 1.11 | 6.60 |
| 5 | 2983 | 3181 | 3008 | 3022 | 1 | 2 | 1.11 | 6.61 |
| 6 | 2983 | 3179 | 3006 | 3020 | 27 | 74 | 1.09 | 6.50 |
| 7 | 2948 | 3119 | 2949 | 2963 | 12 | 33 | 1.06 | 6.07 |
| 8 | 2948 | 3117 | 2947 | 2961 | 20 | 56 | 1.06 | 6.08 |
| 9 | 2933 | 3109 | 2940 | 2953 | 5 | 14 | 1.06 | 6.04 |
| 10 | 1568 | 1680 | 1589 | 1596 | 3 | 7 | 6.55 | 10.90 |
| 11 | 1447 | 1500 | 1418 | 1425 | 6 | 17 | 1.09 | 1.44 |
| 12 | 1439 | 1486 | 1406 | 1412 | 14 | 38 | 1.11 | 1.44 |
| 13 | 1418 | 1474 | 1394 | 1400 | 7 | 19 | 1.09 | 1.40 |
| 14 | 1318 | 1369 | 1294 | 1300 | 17 | 47 | 2.73 | 3.02 |
| 15 | 1310 | 1360 | 1286 | 1292 | 4 | 12 | 1.83 | 2.00 |
| 16 | 1259 | 1331 | 1259 | 1265 | 0 | 0 | 1.60 | 1.67 |
| 17 | 1246 | 1303 | 1232 | 1238 | 4 | 12 | 1.40 | 1.40 |
| 18 | 1218 | 1287 | 1217 | 1222 | 11 | 30 | 1.48 | 1.44 |
| 19 | 1211 | 1263 | 1195 | 1200 | 19 | 53 | 1.69 | 1.59 |
| 20 | 1193 | 1248 | 1180 | 1186 | 4 | 12 | 1.52 | 1.40 |
| 21 | 1189 | 1226 | 1159 | 1165 | 16 | 44 | 1.65 | 1.46 |
| 22 | 1140 | 1219 | 1152 | 1158 | 3 | 9 | 1.42 | 1.25 |
| 23 | 1094 | 1169 | 1106 | 1111 | 5 | 15 | 1.24 | 1.00 |
| 24 | 1068 | 1127 | 1066 | 1071 | 1 | 2 | 1.68 | 1.26 |
| 25 | 1055 | 1107 | 1047 | 1052 | 19 | 52 | 1.76 | 1.27 |
| 26 | 1019 | 1091 | 1032 | 1037 | 7 | 18 | 2.15 | 1.51 |
| 27 | 998 | 1058 | 1001 | 1005 | 17 | 45 | 2.36 | 1.56 |
| 28 | 962 | 1041 | 985 | 989 | 26 | 72 | 2.37 | 1.52 |
| 29 | 934 | 1005 | 950 | 954 | 26 | 71 | 2.25 | 1.34 |
| 30 | 923 | 970 | 917 | 921 | 20 | 54 | 2.72 | 1.51 |
| 31 | 909 | 953 | 901 | 905 | 17 | 47 | 1.91 | 1.02 |
| 32 | 903 | 944 | 893 | 897 | 11 | 31 | 1.56 | 0.82 |
| 33 | 870 | 928 | 878 | 882 | 13 | 34 | 2.15 | 1.09 |
| 34 | 830 | 893 | 844 | 848 | 5 | 15 | 2.14 | 1.00 |
| 35 | 822 | 856 | 810 | 814 | 11 | 29 | 2.67 | 1.15 |
| 36 | 780 | 852 | 806 | 810 | 1 | 2 | 2.94 | 1.26 |
| 37 | 750 | 803 | 759 | 763 | 2 | 6 | 3.33 | 1.27 |
| 38 | 722 | 777 | 735 | 738 | 3 | 9 | 4.81 | 1.71 |
| 39 | 701 | 754 | 713 | 716 | 8 | 22 | 4.79 | 1.60 |
| 40 | 688 | 744 | 703 | 707 | 3 | 9 | 2.88 | 0.94 |
| 41 | 666 | 723 | 684 | 687 | 37 | 100 | 1.95 | 0.60 |
| 42 | 650 | 682 | 645 | 648 | 6 | 16 | 4.07 | 1.11 |
| 43 | 617 | 631 | 597 | 599 | 10 | 26 | 4.57 | 1.07 |
| 44 | 543 | 550 | 520 | 523 | 6 | 16 | 2.92 | 0.52 |
| 45 | - | 513 | 485 | 487 | 3 | 7 | 4.83 | 0.75 |
| 46 | - | 405 | 383 | 385 | 3 | 7 | 2.70 | 0.26 |
| 47 | - | 392 | 371 | 372 | 1 | 3 | 5.18 | 0.47 |
| 48 | - | 350 | 331 | 333 | 2 | 5 | 4.24 | 0.31 |
| 49 | - | 332 | 314 | 315 | 1 | 3 | 3.33 | 0.22 |
| 50 | - | 283 | 268 | 269 | 1 | 3 | 2.40 | 0.11 |
| 51 | - | 266 | 252 | 253 | 2 | 6 | 3.74 | 0.16 |
| 52 | - | 146 | 138 | 138 | 2 | 5 | 5.65 | 0.07 |
| 53 | - | 116 | 110 | 110 | 1 | 2 | 6.48 | 0.05 |
| 54 | - | 77 | 72 | 73 | 2 | 6 | 4.76 | 0.02 |
| r | 0.9998 | 0.9998 | 0.9998 | |||||
| Mean deviation | 80.69 | −0.93 | −71.89 | |||||
| Mean absolute deviation | 80.69 | 14.00 | 71.89 | |||||
| Average absolute error | 5.68 | 1.30 | 5.06 | |||||
| RMSmol | 96.2 | 16.6 | 84.1 | |||||
| RMSover | 86.9 | 14.9 | 75.9 | |||||
| Scaling Factor | 1.0000 | 0.9456 | 0.9500 | |||||
Acknowledgements
References
- Lipshutz, B.H. 5-Membered Heteroaromatic Rings as Intermediates in Organic-Synthesis. Chem. Rev 1986, 86(5), 795–819. [Google Scholar]
- Dean, F.M. Recent advances in furan chemistry I. Adv. Heterocycl. Chem 1982, 30, 167–238. [Google Scholar]
- Dean, F.M.; Sargent, M.V. Furans and Their Benzo Derivatives. In Comprehensive Heterocyclic Chemistry; Bird, C.W., Cheesman, G.W.H., Eds.; Pergamon Press: London, 1984. [Google Scholar]
- Vogel, P.; Cossy, J.; Plumet, J.; Arjona, O. Derivatives of 7-oxabicyclo[2.2.1]heptane in nature and as useful synthetic intermediates. Tetrahedron 1999, 55(48), 13521–13642. [Google Scholar]
- Kappe, C.O.; Murphree, S.S.; Padwa, A. Synthetic applications or furan Diels-Alder chemistry. Tetrahedron 1997, 53(42), 14179–14233. [Google Scholar]
- Grinsteiner, T.J.; Kishi, Y. Synthetic Studies Towards Batrachotoxin.1. A Furan-Based Intramolecular Diels-Alder Route to Construct the a-D Ring-System. Tetrahedron Lett 1994, 35(45), 8333–8336. [Google Scholar]
- Demircan, A.; Parsons, P.J. Preparation of tricyclic nitrogen heterocycle via Intramolecular Diels-Alder reaction. Heterocycl. Commun 2002, 8(6), 531–536. [Google Scholar]
- Demircan, A.; Karaarslan, M.; Turac, E. A facile synthesis of heterotricycles from furfurylbromoalkenes using thermal IMDA cycloaddition. Heterocycl. Commun 2006, 3–4, 233– 240. [Google Scholar]
- Williamson, A.W. XXII.-On etherification. Q. J. Chem. Soc 1852, 4, 229–239. [Google Scholar]
- Adamchuk, K.A.; Parker, K.A. Intramolecular Diels-Alder reactions of the furan diene. Tetrahedron Lett 1978, 19, 1689–1692. [Google Scholar]
- Buyukgungor, O.; Kosar, B.; Demircan, A.; Turac, E. 6-Bromo-8-thia-1,4-epoxybicyclo[4.3.0]non-2-ene. Acta Crystallog E 2005, 61, O1441–O1442. [Google Scholar]
- Arslan, H.; Florke, U.; Kulcu, N. Theoretical studies of molecular structure and vibrational spectra of O-ethyl benzoylthiocarbamate. Spectrochim. Acta A 2007, 67(3–4), 936–943. [Google Scholar]
- Arslan, H.; Algül, Ö.; Dündar, Y. Structural and spectral studies on 3-(6-benzoyl-5-chloro-2-benzoxazolinon-3-yl) propanoic acid. Vib. Spectrosc 2007, 44, 248–255. [Google Scholar]
- Arslan, H.; Emen, F.M.; Kulcu, N. Structure and vibrational spectra of N,N-dimethyl-N′-(2-chlorobenzoyl)thiourea: Hartree-Fock and density functional theory studies. Asian J. Chem 2007, 19, 1888–1896. [Google Scholar]
- Arslan, H.; Algul, O. Theoretical studies of molecular structure and vibrational spectra of 2-ethyl-1H-benzo[d]imidazole. Asian J. Chem 2007, 19, 2229–2235. [Google Scholar]
- Arslan, H.; Algül, Ö. Synthesis and Ab Initio/DFT Studies on 2-(4-methoxyphenyl) benzo[d]thiazole. Int. J. Mol. Sci 2007, 8, 760–776. [Google Scholar]
- Arslan, H.; Flörke, U.; Külcü, N.; Binzet, G. The molecular structure and vibrational spectra of 2-chloro-N-(diethylcarbamothioyl)benzamide by Hartree–Fock and density functional methods. Spectrochim. Acta A 2007. [Google Scholar] [CrossRef]
- Arslan, H.; Algul, Ö.; Önkol, T. Vibrational spectroscopy investigation using ab-initio and density functional theory analysis on the structure of 3-(6-benzoyl-2-oxobenzo[d]oxazol-3(2H)-yl)propanoic acid. Spectrochim. Acta A 2007. [Google Scholar] [CrossRef]
- Arslan, H.; Demircan, A.; Göktürk, E. Vibrational spectroscopy investigation using ab initio and density functional theory analysis on the structure of 5-chloro-10-oxa-3-thiatricyclo[ 5.2.1.01,5]dec-8-ene-3,3-dioxide. Spectrochim. Acta A 2007. [Google Scholar] [CrossRef]
- Arslan, H.; Algül, Ö. Vibrational spectrum and assignments of 2-(4-methoxyphenyl)-1H-benzo[d]imidazole by ab initio Hartree–Fock and density functional methods. Spectrochim. Acta A 2007. [Google Scholar] [CrossRef]
- Kupka, T.; Wrzalik, R.; Pasterna, G.; Pasterny, K. Theoretical DFT and experimental Raman and NMR studies on thiophene, 3-methylthiophene and selenophene. J. Mol. Struct 2002, 616, 17–32. [Google Scholar]
- Wysokinski, R.; Kuduk-Jaworska, J.; Michalska, D. Electronic structure, Raman and infrared spectra, and vibrational assignment of carboplatin. Density functional theory studies. J. Mol. Struc-Theochem 2006, 758, 169–179. [Google Scholar]
- Kuppens, T.; Vandyck, K.; VanderEycken, J.; Herrebout, W.; VanderVeken, B.; Bultinck, P.A. DFT conformational analysis and VCD study on methyl tetrahydrofuran-2-carboxylate. Spectrochim. Acta A 2007, 67, 402–411. [Google Scholar]
- Hanuza, J.; Sasiadek, W.; Michalski, J.; Lorenc, J.; Maczka, M.; Kaminskii, A.A.; Butashin, A.V.; Klapper, H.; Hulliger, J.; Mohmed, A.F.A. Polarized Raman and infrared spectra of the salol crystal - chemical quantum calculations of the vibrational normal modes. Vib. Spectrosc 2004, 34, 253–268. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc 2007. [Google Scholar] [CrossRef]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Exchange-correlation functional with broad accuracy for metallic and nonmetallic compounds, kinetics, and noncovalent interactions. J. Chem. Phys 2005, 123. [Google Scholar]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory. Comput 2006, 2(2), 364–382. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys 2006, 125. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; Iyengar, S.S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G.A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J.E.; Hratchian, H.P.; Cross, J.B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R.E.; Yazyev, O.; Austin, A.J.; Cammi, R.; Pomelli, C.; Ochterski, J.W.; Ayala, P.Y.; Morokuma, K.; Voth, G.A.; Salvador, P.; Dannenberg, J.J.; Zakrzewski, V.G.; Dapprich, S.; Daniels, A.D.; Strain, M.C.; Farkas, O.; Malick, D.K.; Rabuck, A.D.; Raghavachari, K.; Foresman, J.B.; Ortiz, J.V.; Cui, Q.; Baboul, A.G.; Clifford, S.; Cioslowski, J.; Stefanov, B.B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R.L.; Fox, D.J.; Keith, T.; Al-Laham, M.A.; Peng, C.Y.; Nanayakkara, A.; Challacombe, M.; Gill, P.M.W.; Johnson, B.; Chen, W.; Wong, M.W.; Gonzalez, C.; Pople, J.A. Gaussian 03, Revision C.02; Gaussian, Inc: Wallingford CT, 2004. [Google Scholar]
- Moller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev 1934, 46, 618–622. [Google Scholar]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Phys. Rev. A 1988, 38(6), 3098–3100. [Google Scholar]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys. Rev. B 1988, 37(2), 785–789. [Google Scholar]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys 1998, 108(2), 664–675. [Google Scholar]
- Burke, K.; Perdew, J.P.; Wang, Y. Electronic Density Functional Theory: Recent Progress and New Directions; Dobson, J.F., Vignale, G., Das, M.P., Eds.; Plenum: New York, 1998. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry .3. The Role of Exact Exchange. J. Chem. Phys 1993, 98(7), 5648–5652. [Google Scholar]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar]
- Foresman, J.B.; Frisch, E. Exploring Chemistry with Electronic Structure Methods: A Guide to Using Gaussian; Gaussian: Pitttsburg, PA, 1993. [Google Scholar]
- Scott, A.P.; Radom, L. Harmonic vibrational frequencies: An evaluation of Hartree-Fock, Moller-Plesset, quadratic configuration interaction, density functional theory, and semiempirical scale factors. J. Phys. Chem.-US 1996, 100, 16502–16513. [Google Scholar]
- Arslan, H. Performance Analysis of Vibrational Frequencies, PAVF 1.0; Mersin: Turkey, 2007. [Google Scholar]
- GaussView, Version 3.07; Dennington, Roy, II; Keith, Todd; Millam, John; Eppinnett, Ken; Hovell, W Lee; Gilliland, Ray. Semichem, Inc: Shawnee Mission, KS, 2003. [Google Scholar]
- Jamróz, M.H. Vibrational Energy Distribution Analysis VEDA 4; Warsaw, 2004. [Google Scholar]
- Supuran, C.T.; Casini, A.; Scozzafava, A. Protease inhibitors of the sulfonamide type: Anticancer, antiinflammatory, and antiviral agents. Med. Res. Rev 2003, 23, 535–558. [Google Scholar]
- Palmer, J.T.; Rasnick, D.; Klaus, J.L.; Bromme, D. Vinyl Sulfones as Mechanism-Based Cysteine Protease Inhibitors. J. Med. Chem 1995, 38, 3193–3196. [Google Scholar]
- Roush, W.R.; Gwaltney, S.L.; Cheng, J.M.; Scheidt, K.A.; McKerrow, J.H.; Hansell, E. Vinyl sulfonate esters and vinyl sulfonamides: Potent, irreversible inhibitors of cysteine proteases. J. Am. Chem. Soc 1998, 120, 10994–10995. [Google Scholar]
- Simpkins, N.S. The Chemistry of Vinyl Sulfones. Tetrahedron 1990, 46, 6951–6984. [Google Scholar]
- Kosar, B.; Gokturk, E.; Demircan, A.; Buyukgungor, O. 5-bromo-10-oxa-3-thiatricyclo[5.2.1.0(1,5)]-dec-8-ene 3,3-dioxide. Acta Cryst. E 2006, 62, O3868–O3869. [Google Scholar]
- Cioslowski, J. A New Population Analysis Based on Atomic Polar Tensors. J. Am. Chem. Soc 1989, 111, 8333–8336. [Google Scholar]
- Visscher, L.; Dyall, K.G. Relativistic and correlation effects on molecular properties.1. The dihalogens F-2, Cl-2, Br-2, I-2, and At-2. J. Chem. Phys 1996, 104(22), 9040–9046. [Google Scholar]
- Xiao, C.Y.; Kruger, S.; Belling, T.; Mayer, M.; Rosch, N. Relativistic effects on geometry and electronic structure of small Pd-n species (n=1, 2, 4). Int. J. Quant. Chem 1999, 74(4), 405–416. [Google Scholar]
- Bauschlicher, C.W.; Langhoff, S.R. The calculation of accurate harmonic frequencies of large molecules: The polycyclic aromatic hydrocarbons, a case study. Spectrochim. Acta A 1997, 53(8), 1225–1240. [Google Scholar]
- Bayari, S.; Saglam, S.; Ustundag, H.F. Experimental and theoretical studies of the vibrational spectrum of 5-hydroxytryptamine. J. Mol. Struc.-Theochem 2005, 726(1–3), 225–232. [Google Scholar]
- Silverstein, M.; Basseler, C.G.; Morill, C. Spectrometric Identification of Organic Compounds; Wiley: New York, 1981. [Google Scholar]
- Kimmelma, R.; Hotokka, M. Structure-stability relationships in unsaturated sulfur compounds VI. An ab initio study of the stable conformations of (E)- and (Z)-2-methylthio-, methylsulfinyl-and methylsulfonyl-2-butenes. J. Mol. Struct.-Theochem 1997, 418(2–3), 89–196. [Google Scholar]
- Sundaraganesan, N.; Meganathan, C.; Anand, B.; Lapouge, C. FT-IR, FT-Raman spectra and ab initio DFT vibrational analysis of p-bromophenoxyacetic acid. Spectrochim. Acta A 2007, 66(3), 773–780. [Google Scholar]
- Cabral, O.V.; Tellez, C.A.; Giannerini, T.; Felcman, J. Fourier-transform infrared spectrum of aspartate hydroxo-aqua nickel(II) complex and DFT-B3LYP/3-21G and 6-311G structural and vibrational calculations. Spectrochim. Acta A 2005, 61(1–2), 337–345. [Google Scholar]
- Bensebaa, F.; Zhou, Y.; Brolo, A.G.; Irish, D.E.; Deslandes, Y.; Kruus, E.; Ellis, T.H. Raman characterization of metal-alkanethiolates. Spectrochim. Acta A 1999, 55(6), 1229–1236. [Google Scholar]
- Mohan, J. Organic Spectroscopy Principles and Applications; Narosa Publishing House: New Delhi, 2001. [Google Scholar]
- Gunasekaran, S.; Kumar, R.T.; Ponnusamy, S. Vibrational spectra and normal coordinate analysis of diazepam, phenytoin and phenobarbitone. Spectrochim. Acta A 2006, 65, 1041–1052. [Google Scholar]
© 2007 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Arslan, H.; Demircan, A. The Molecular Structure and Vibrational Spectrum of 6-bromo-8-thia-1,4-epoxybicyclo[4.3.0]non-2-ene. Int. J. Mol. Sci. 2007, 8, 1064-1082. https://doi.org/10.3390/i8111064
Arslan H, Demircan A. The Molecular Structure and Vibrational Spectrum of 6-bromo-8-thia-1,4-epoxybicyclo[4.3.0]non-2-ene. International Journal of Molecular Sciences. 2007; 8(11):1064-1082. https://doi.org/10.3390/i8111064
Chicago/Turabian StyleArslan, Hakan, and Aydın Demircan. 2007. "The Molecular Structure and Vibrational Spectrum of 6-bromo-8-thia-1,4-epoxybicyclo[4.3.0]non-2-ene" International Journal of Molecular Sciences 8, no. 11: 1064-1082. https://doi.org/10.3390/i8111064
APA StyleArslan, H., & Demircan, A. (2007). The Molecular Structure and Vibrational Spectrum of 6-bromo-8-thia-1,4-epoxybicyclo[4.3.0]non-2-ene. International Journal of Molecular Sciences, 8(11), 1064-1082. https://doi.org/10.3390/i8111064