Crystallization and Characterization of Galdieria sulphuraria RUBISCO in Two Crystal Forms: Structural Phase Transition Observed in P21 Crystal Form

Abstract
:1. Introduction
2. Results and Discussion
2.1 Structure solution and the architecture
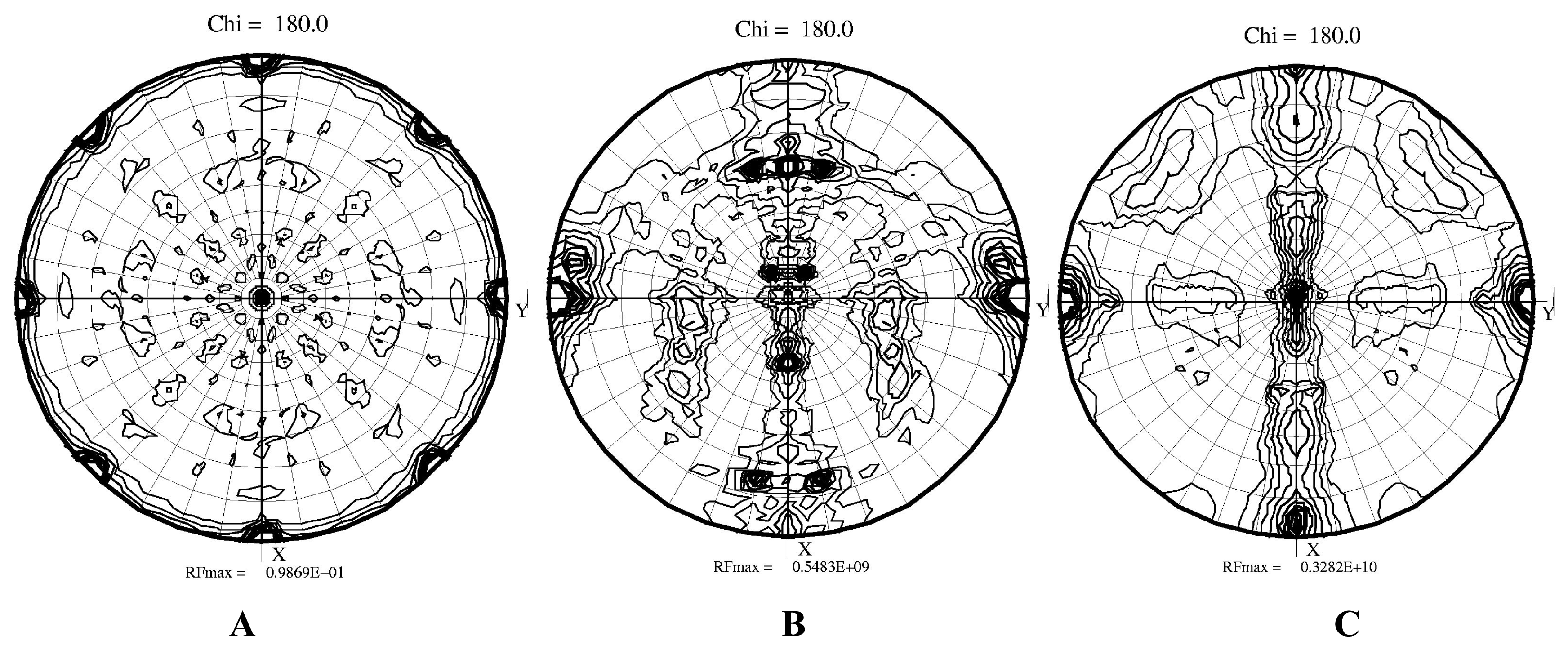
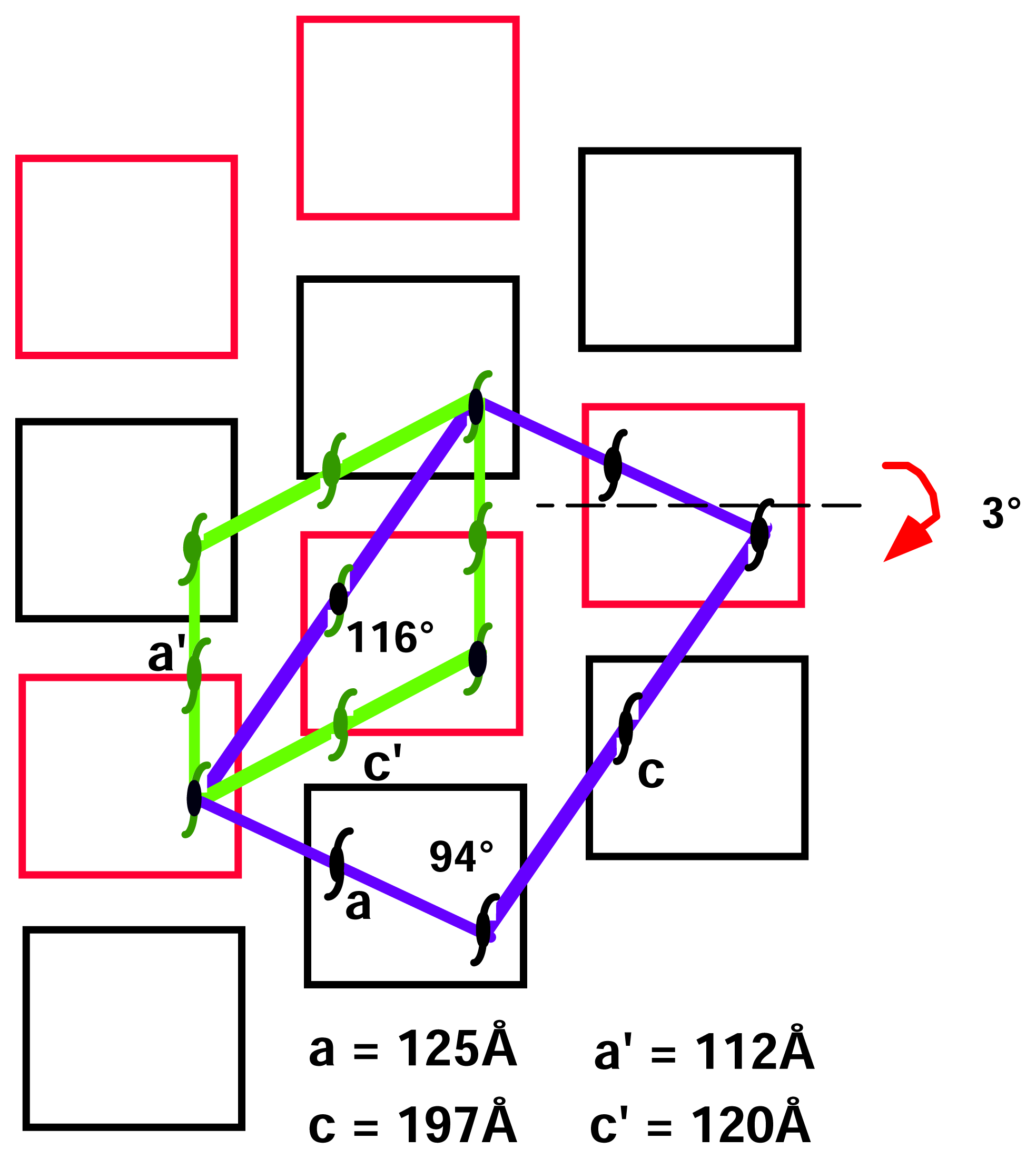
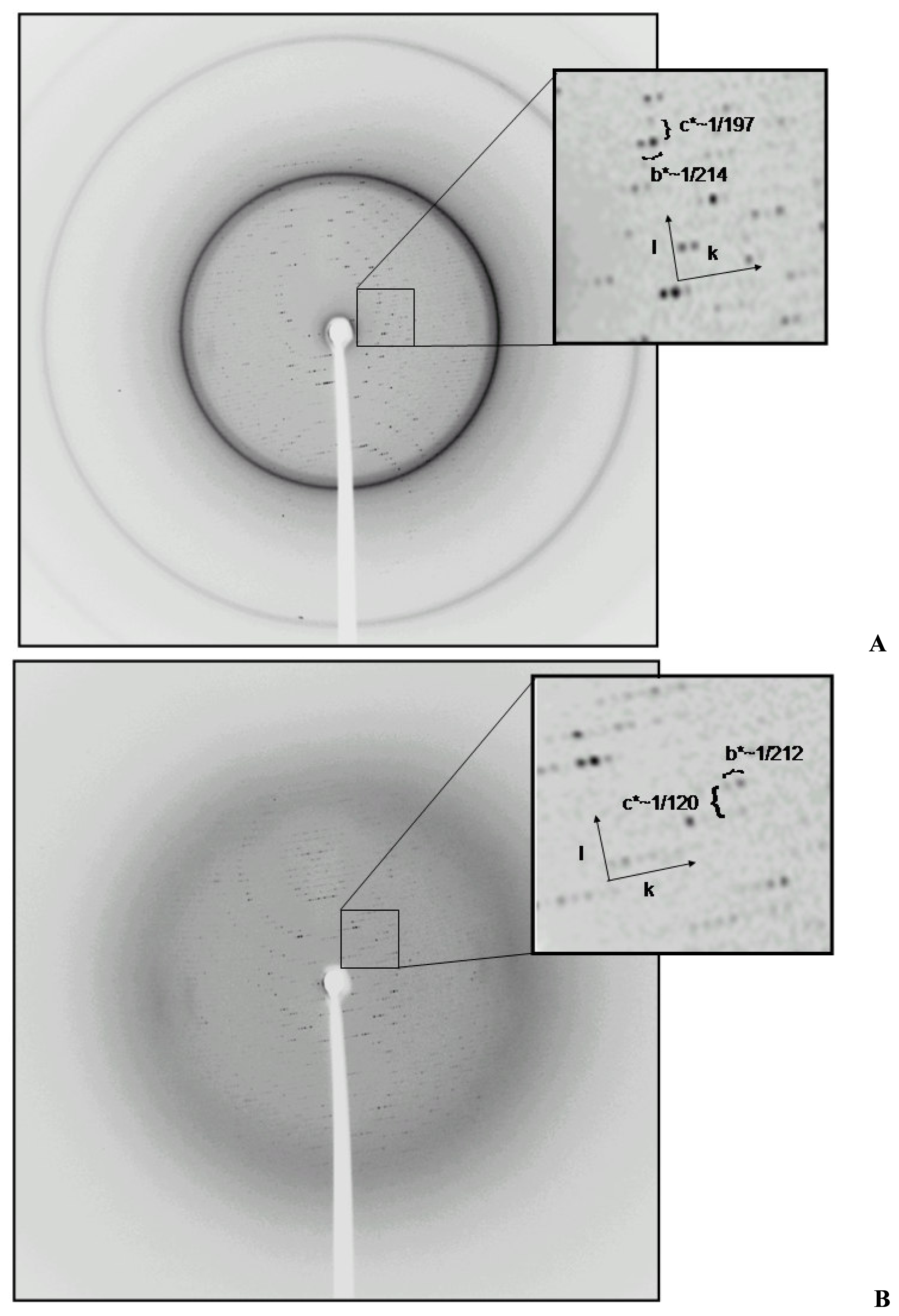
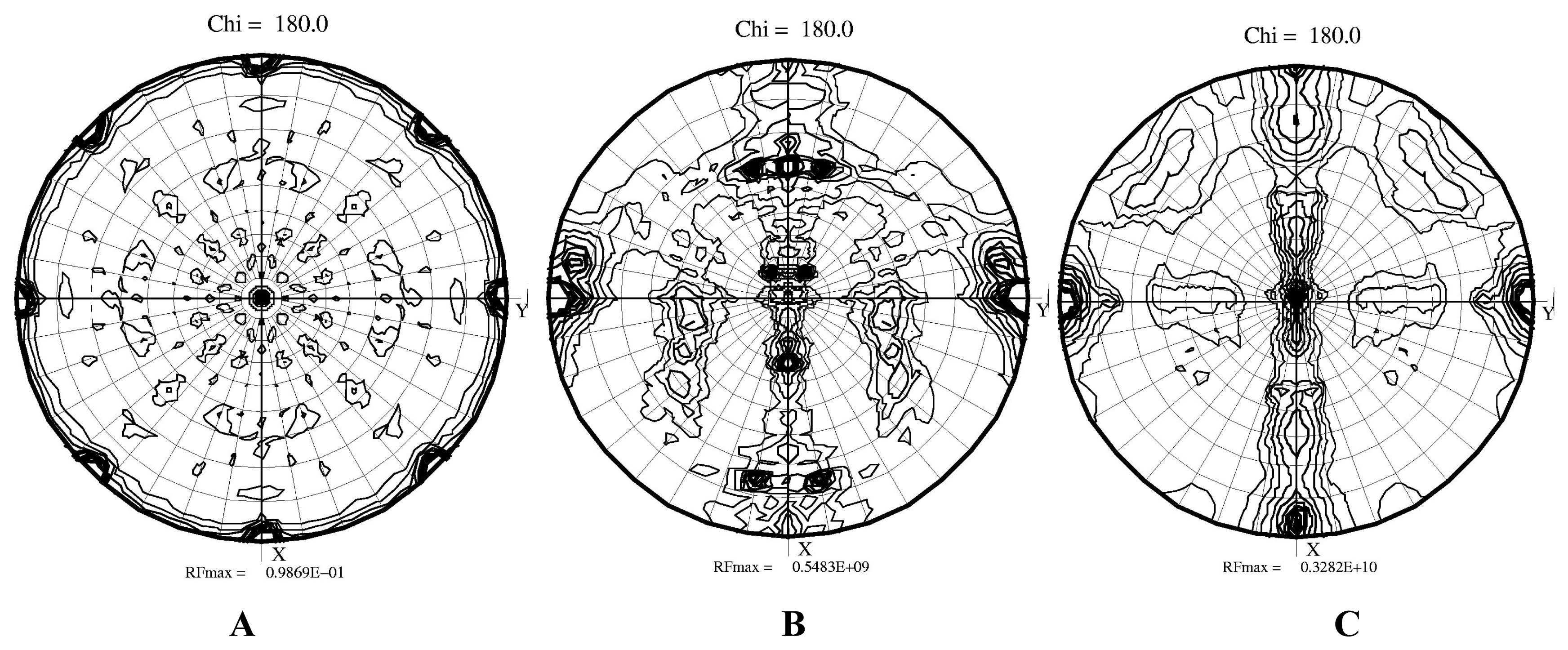
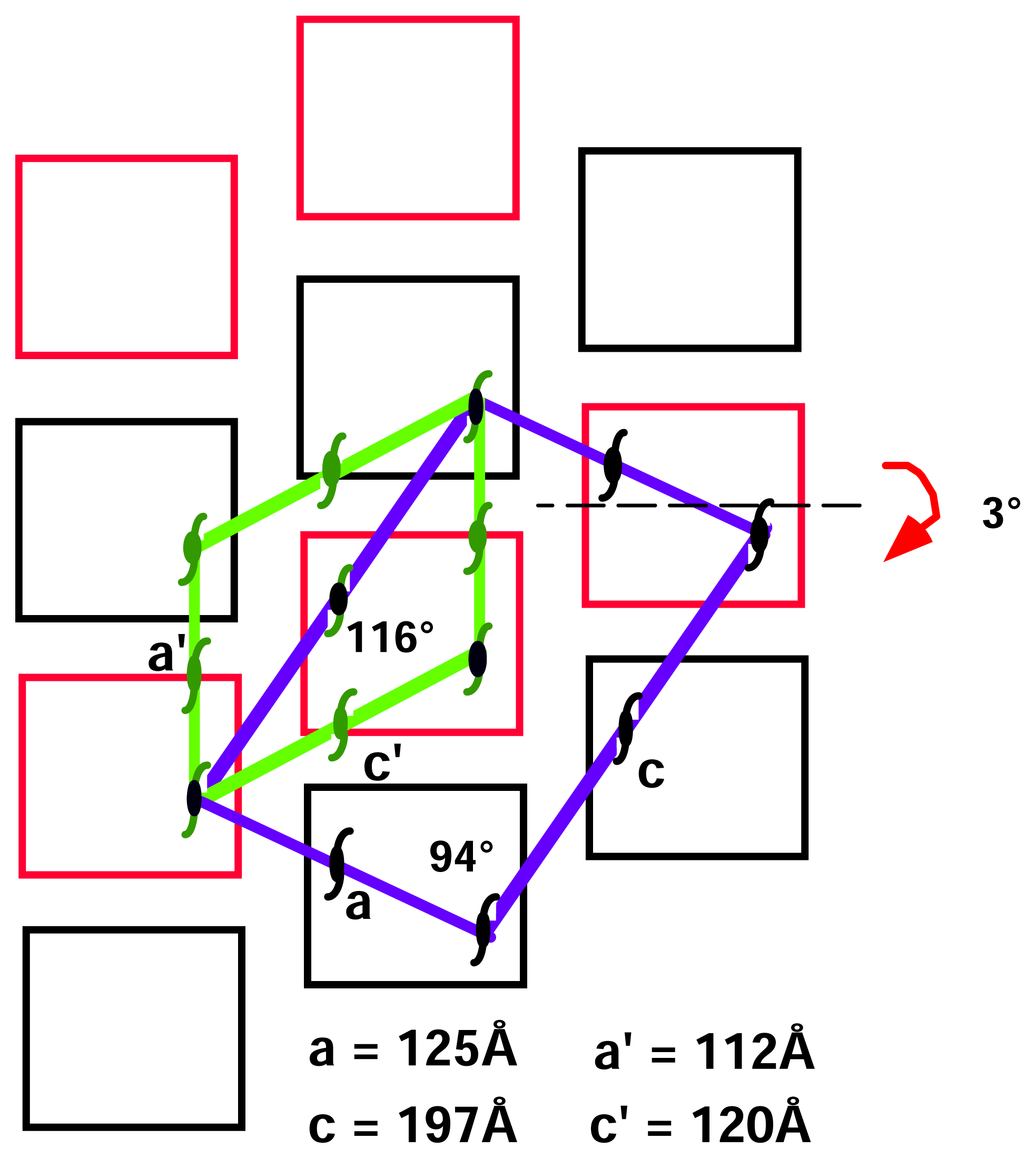
2.2 Structural phase transition in the P21 crystal form
3. Experimental Section
3.1 Materials
3.2 Cell Growth
3.3 Purification
3.4 Crystallization
3.5 Data Collection
3.6 Molecular replacement

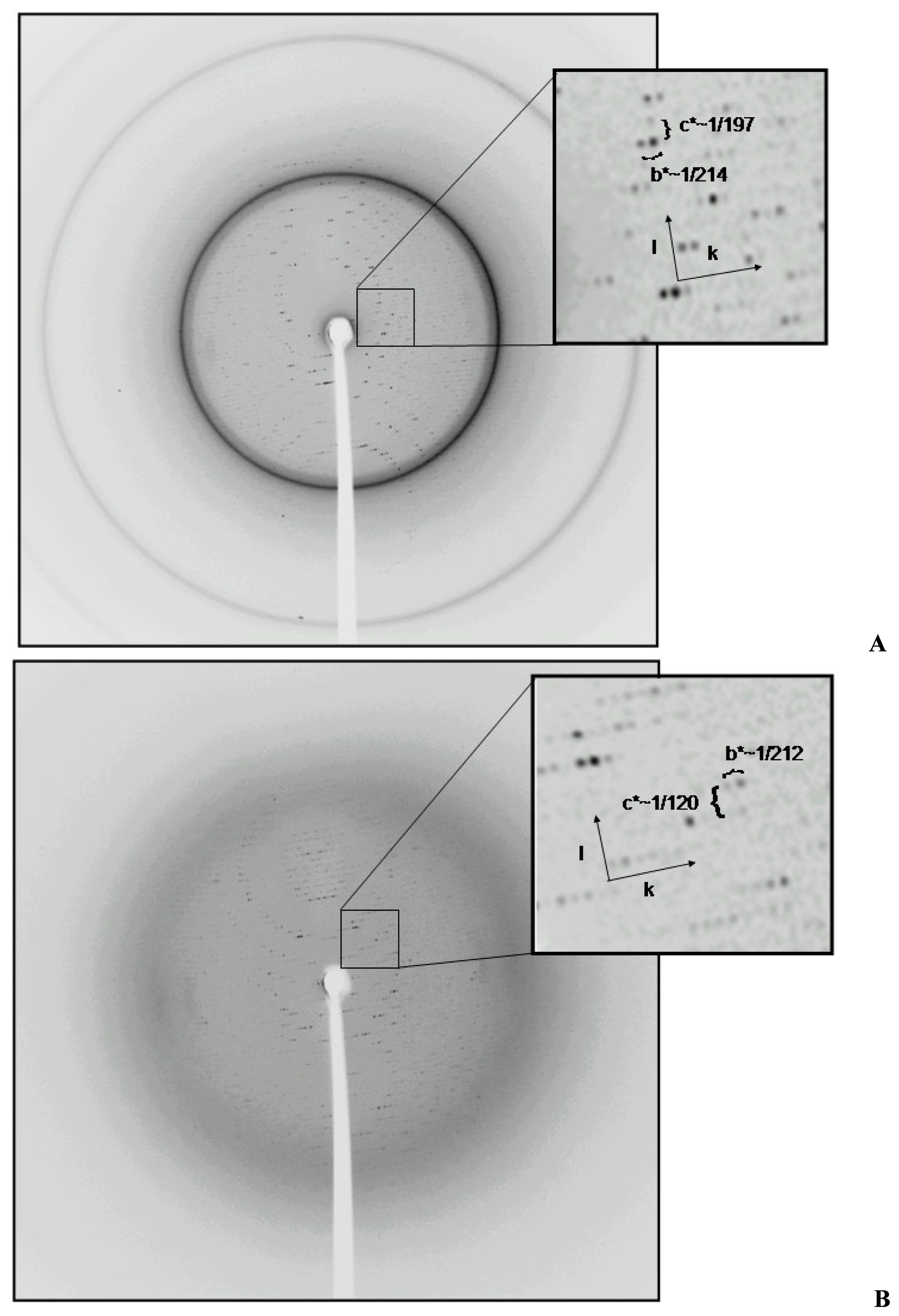



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal form | 1 | 2a | 2b |
|---|---|---|---|
| Space group | I422 | P21 | P21 |
| Cell dimensions (Å) | |||
| a | 136.9, | 124.7, | 111.8, |
| b | 136.9, | 214.6, | 212.3, |
| c | 122.9, | 197.5, | 120.4, |
| β | 90.0 | 94.7 | 116.9 |
| Resolution (Å) | 37-2.0 | 34-2.4 | 34-2.8 |
| Rmerge* | 0.075(0.41) | 0.114(0.52) | 0.146(0.39) |
| I/σ(I) | 7.2(2.1) | 4.2(1.8) | 3.7(1.9) |
| Redundancy | 6.1(3.1) | 2.3(1.7) | 1.9(1.7) |
| No. reflections | 37654 | 269663 | 99767 |
| Completness (%) | 89.3(81.0) | 85.7(83.2) | 79.7(79.2) |
| No. atoms placed in the asymmetric unit | 6224 | 74224 | 37112 |
Acknowledgements
References and Notes
- Zelitch, I. Pathways of carbon fixation in green plants. Ann. Rev. Biochem 1975, 44, 123–45. [Google Scholar]
- Pereto, J. G.; Velasco, A. M.; Becerra, A.; Lazcano, A. Comparative biochemistry of CO2 fixation and the evolution of autotrophy. Int. Microbiol 1999, 2, 3–10. [Google Scholar]
- Newman, J.; Gutteridge, S. Structure of an effector-induced inactivated state of ribulose 1,5- bisphosphate carboxylase/oxygenase: the binary complex between enzyme and xylulose 1,5- bisphosphate. Structure 1994, 2, 495–502. [Google Scholar]
- Andersson, I. Large structures at high resolution: the 1.6 A crystal structure of spinach ribulose- 1,5-bisphosphate carboxylase/oxygenase complexed with 2-carboxyarabinitol bisphosphate. J. Mol. Biol 1996, 259, 160–74. [Google Scholar]
- Zhang, K.Y.; Cascio, D.; Eisenberg, D. Crystal structure of the unactivated ribulose 1,5- bisphosphate carboxylase/oxygenase complexed with a transition state analog, 2-carboxy-D-arabinitol 1,5-bisphosphate. Protein Sci 1994, 3, 64–9. [Google Scholar]
- Sugawara, H.; Yamamoto, H.; Shibata, N.; Inoue, T.; Okada, S.; Miyake, C.; Yokota, A.; Kai, Y. Crystal structure of carboxylase reaction-oriented ribulose 1,5-bisphosphate carboxylase/oxygenase from a thermophilic red alga, Galdieria partita. J. Biol. Chem 1999, 274, 15655–15661. [Google Scholar]
- Cleland, W.W.; Andrews, T.J.; Gutteridge, S.; Hartman, F.C.; Lorimer, G.H. Mechanism of Rubisco: The carbamate as general base. Chem. Rev 1998, 98, 549–561. [Google Scholar]
- Zhang, N.; Portis, A.R., Jr. Mechanism of light regulation of Rubisco: a specific role for the larger Rubisco activase isoform involving reductive activation by thioredoxin-f. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 9438–43. [Google Scholar]
- Sekbach, J.; Fredrick, J.F. A primaeval alga bridging the blue-green and the red algae: further biochemical and ultrastructure studies of Cyanidium caldarium with special reference to the plastid membranes. Microbios 1980, 29, 135–47. [Google Scholar]
- Valentin, K.; Zetsche, K. Structure of the Rubisco operon from the unicellular red alga Cyanidium caldarium: evidence for a polyphyletic origin of the plastids. Mol. Gen. Genet 1990, 222, 425–30. [Google Scholar]
- Spreitzer, R. J.; Salvucci, M. E. Rubisco: structure, regulatory interactions, and possibilities for a better enzyme. Ann. Rev. Plant. Biol 2002, 53, 449–75. [Google Scholar]
- Uemura, K.; Anwaruzzaman, M.; Miyachi, S.; Yokota, A. Ribulose-1,5-bisphosphate carboxylase/oxygenase from thermophilic red algae with a strong specificity for CO2 fixation. Biochem. Biophys. Res. Commun 1997, 233, 568–71. [Google Scholar]
- Ford, T. W. Ribulose 1,5-bisphosphate carboxylase from the thermophilic, acidophilic alga, Cyanidium caldarium (Geitler). Purification, characterization and thermostability of the enzyme. Biochem. Biophys. Acta 1979, 569, 239–248. [Google Scholar]
- Curmi, P.M.; Cascio, D.; Sweet, R.M.; Eisenberg, D.; Schreuder, H. Crystal structure of the unactivated form of ribulose-1,5-bisphosphate carboxylase/oxygenase from tobacco refined at 2.0- A resolution. J. Biol. Chem 1992, 267, 16980–9. [Google Scholar]
- Okano, Y.; Mizohata, E.; Xie, Y.; Matsumura, H.; Sugawara, H.; Inoue, T.; Yokota, A.; Kai, Y. X-ray structure of Galdieria Rubisco complexed with one sulfate ion per active site. FEBS Lett 2002, 527, 33–6. [Google Scholar]
- Harata, K.; Akiba, T. Phase transition of triclinic hen egg-white lysozyme crystal associated with sodium binding. Acta Crystallogr. D 2004, 60, 630–7. [Google Scholar]
- Weiss, M. S.; Hilgenfeld, R. Dehydration leads to a phase transition in monoclinic factor XIII crystals. Acta Crystallogr D 1999, 55, 1858–62. [Google Scholar]
- Declerq, J.P.; Evrard, C. A twinned monoclinic crystal form of human peroxiredoxin 5 with eight molecules in the asymmetric unit. Acta Crystallogr. D 2001, 57, 1829–1835. [Google Scholar]
- Dauter, Z.; Li, M.; Wlodawer, A. Practical experience with the use of halides for phasing macromolecular structures: a powerful tool for structural genomics. Acta Crystallogr. D 2001, 57, 239–249. [Google Scholar]
- Skrzypczak-Jankun, E.; Bianchet, M. A.; Amzel, M. L.; Funk, M. O., Jr. Flash-freezing causes a stress-induced modulation in a crystal structure of soybean lipoxygenase. Acta Crystallogr. D 1996, 52, 959–65. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode. Method. Enzymol 1997, 276, 307–326. [Google Scholar]
- Brunger, A. T.; Adams, P. D.; Clore, G. M.; DeLano, W. L.; Gros, P.; Grosse-Kunstleve, R. W.; Jiang, J. S.; Kuszewski, J.; Nilges, M.; Pannu, N. S.; Read, R. J.; Rice, L. M.; Simonson, T.; Warren, G. L. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 1998, 54, 905–21. [Google Scholar]
© 2007 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
Baranowski, M.; Stec, B. Crystallization and Characterization of Galdieria sulphuraria RUBISCO in Two Crystal Forms: Structural Phase Transition Observed in P21 Crystal Form. Int. J. Mol. Sci. 2007, 8, 1039-1051. https://doi.org/10.3390/i8101039
Baranowski M, Stec B. Crystallization and Characterization of Galdieria sulphuraria RUBISCO in Two Crystal Forms: Structural Phase Transition Observed in P21 Crystal Form. International Journal of Molecular Sciences. 2007; 8(10):1039-1051. https://doi.org/10.3390/i8101039
Chicago/Turabian StyleBaranowski, Michael, and Boguslaw Stec. 2007. "Crystallization and Characterization of Galdieria sulphuraria RUBISCO in Two Crystal Forms: Structural Phase Transition Observed in P21 Crystal Form" International Journal of Molecular Sciences 8, no. 10: 1039-1051. https://doi.org/10.3390/i8101039
APA StyleBaranowski, M., & Stec, B. (2007). Crystallization and Characterization of Galdieria sulphuraria RUBISCO in Two Crystal Forms: Structural Phase Transition Observed in P21 Crystal Form. International Journal of Molecular Sciences, 8(10), 1039-1051. https://doi.org/10.3390/i8101039