I. Introduction
Single reference coupled cluster theory, [
1,
2] especially CCSD(T) [
3,
4], is a highly successful approach to describe the electronic structure of systems that are qualitatively reasonably well described by a single determinant (many closed-shell molecules and high-spin open-shell systems) [
5,
6,
7,
8]. Likewise coupled cluster response theory (CCLRT [
9,
10,
11]) and its cousin equation-of-motion coupled cluster theory (EOM-CC [
5,
12]) are well suited to describe singly excited states and radicals. An important prerequisite for the applicability of the CC response approach is the presence of a nearby state that can serve as the reference in the parent state CCSD calculation. Most commonly the reference state is a closed-shell system that differs by up to one electron from the states of interest. The standard CCSD equations are solved for the reference state and the primary role of the cluster operator
is to include dynamical correlation, which tends to be transferable from one state to the other. The electronic states of interest are obtained by diagonalizing the transformed Hamiltonian
over the proper configurations. In practice, if an excited state is dominated by
1p1h (i.e. single) excitations (or
1h, or
1p substitutions in case of IP-EOMCC and EA-EOMCC respectively [
13,
14,
15,
16]) one needs to include one more excitation level in the diagonalization space to achieve an accuracy of about 0.1 - 0.3 eV [
17,
18]. The accuracy of EOMCC and/or CCLRT can be further improved by including perturbative triple corrections (of the iterative or non-iterative type) [
19,
20,
21,
22,
23], while also full EOM-CCSDT results have been presented [
24,
25]. Recently, Krylov [
26,
27] pushed the EOM strategy a level further by choosing as the reference state a high spin (usually triplet) state and diagonalizing over a manifold of states of different spin multiplicity (the so-called spin-flip EOM method). If the dominant part of the final state wave function resides in the principal sector (related to the reference by a single spin-flip excitation) the approach has been found to yield promising results. The (triplet based) spin flip EOM approach provides access to biradicals and the breaking of single bonds and appears a nice extension of the tools in the quantum chemistry community.
Other CC variants exist to describe excited states that involve yet another similarity transform of the Hamiltonian related to ionization potentials and electron affinities. The purpose of the second transformation is to remove (or decrease) the coupling of the primary sector to the remaining sectors [
28,
29,
30,
31]. Fock Space Coupled Cluster [
32,
33,
34,
35] and Similarity Transformed EOMCC (STEOM [
30,
36,
37,
38]) are the best known variants of such an approach and they provide a very efficient approach to excitation spectra as the final diagonalization space can be very compact. Moreover, these approaches have traditionally also been used to describe certain multireference situations. A recent example is the description of the electronic states of the NO
3 cation [
39], most of which have significant multireference character. The NO
3- anion is well described by CCSD and this is the state that is used as a reference, while the final diagonalization is over states that remove two electrons from the reference, e.g.
. For this reason the method is referred to as the double-ionization potential STEOM (DIP-STEOM) approach. Beyond computational efficiency a strong feature of the FSCC and STEOM approaches is the fact that they are rigorously spin-adapted and size-consistent. A weakness is that the reference state may be far removed from the actual states of interest and orbital relaxation effects may be substantial which are difficult to describe through configuration mixing. The reference state can become entirely artificial in these schemes as exemplified by results for the vibrational frequencies of ozone using the DIP-STEOM approach based on the di-anion of
O3
[
30]. In reality the dianion of ozone is non-existent and the bound reference state is an artifact of the basis set. Therefore in such cases the DIP approach can
only give reasonable (and even excellent) results in
small basis sets, lacking diffuse character in particular.
The idea behind all of these approaches is that information on dynamical correlation coefficients are obtained from states that are well described by single reference theories, and these are then transferred to the actual states of interest (through similarity transformations [
28,
29,
30,
31]). The general success of these approaches indicates that dynamical correlation is largely transferable (the concept of valence universality, e.g. [
34,
40]), and it also tells us that the parameterizations for the wave functions can be quite compact. All of the above approaches can be viewed as internally contracted methodologies [
41}: The cluster operator acts upon the qualitatively correct wave function as a whole. However, the fact that in practice the cluster coefficients
have to be obtained from a well-behaved single reference state is a limitation. Such a state is not always present, or its presence may depend on the nuclear geometry considered, while the transferability of dynamical correlation will depend on the similarity of reference and target states. These statements are equivalent to the fact that valence universal (or Fock Space) theories as discussed in detail by Mukherjee and Pal [
34], and Paldus and Jeziorski [
40] have their limitations for the general open-shell problem. The
parameterizations are very suitable in my mind, but the equations to determine the amplitudes, i.e. the valence universality concept may break down.
In this paper we investigate simple multireference situations that can be characterized by the fact that one virtual orbital dominates the correlation effects. Two simple diatomic molecules, O
2 and F
2, are used as examples, and they share the property that qualitatively correct wave functions (for ground and valence excited states) can be described by creating two holes in a formal closed-shell di-anion determinant. Therefore our basic formulation follows a double ionization type of strategy. We will use the same parameterization for the wave function as is used in the double ionization variant of equation-of-motion coupled cluster theory (DIP-EOMCC [
39]). The
equations that determine these parameters are quite different however. In principle
all parameters are determined specifically for the state of interest. We will discuss a number of slightly different variants and generalize the state-specific scheme such that coefficients can be determined in a state-averaged way. Finally we come full circle and investigate if the state specific coefficients for
one particular state could also be used for other states, implying that they would again be transferable or valence universal. The approaches we discuss in this paper are steps towards a completely general internally contracted multireference coupled cluster approach along the lines discussed in [
29,
42,
43]. However, none of the approaches we discuss here are rigorously size-consistent, in the sense that an open-shell system does not separate quite correctly into open-shell subsystems. For this reason we term our approaches state-selective variants of equation-of-motion coupled cluster theory, rather than multireference CC. The methods are size-intensive in the sense that the energies scale properly if closed-shell systems are added at infinity.
II. Theory
In this paper we consider wave functions that are qualitatively well described by the parameterization
The closed-shell determinant
contains two more electrons than the state(s) of interest and serves as the
vacuum in the many-body theory. Occupied orbitals in
are indicated as
i,
j,
k,
l etc., while virtual orbitals carry labels
a,
b,
c,
d. The state
is the
reference state and is assumed to contain the dominant configurations in the target wave function. The orbitals in
are defined such that the reference state is optimal in some sense. The orbitals can be defined for example using an MCSCF procedure. Let us note here that our reference state has a slightly different character than in other multireference approaches, as there are no active space restrictions in Eqn. (1). Rather, the 'active' space in our formulation is implicit by the definition of the vacuum state that contains one more spatial orbital than the number of electron pairs. Once the closed-shell vacuum determinant is defined all orbitals are treated equivalently.
The complete correlated wave function is parameterized as
where
is the traditional closed-shell CCSD operator, where particles and holes are defined with respect to
. The linear operator
has the form
such that a complete operator
(in a full CI sense containing up to (
N + 2)
h −
Np excitations) can describe any state with 2 less electrons than
. Substituting the parameterization in the Schrödinger equation and multiplying by
we find
The operator
is hence the eigenvector of a transformed Hamiltonian, as in EOMCC. In principle the transformation with
can be arbitrary, and its purpose is to improve the convergence properties of the
truncated diagonalization, that we wish to carry only to the
3h1p level. The arbitrariness or complete redundancy of the operator
in the exact case (i.e. full CI expansion of
) indicates that a proper definition of the equations for
is not straightforward. It also implies that different values for
could lead to identical forms for the wave functions if the operator
is adjusted accordingly. This arbitrariness/redundancy is at the heart of the difficulty of formulating a multireference CC theory. Formal theoretical considerations can help only so much. Actual computational schemes have to be tried out in practice and their robustness and general applicability have to be established by elaborate testing.
While the proper equations for
require further discussion (see below) the equations for the operator
, truncated as in Eqn. (4) are clear-cut. We will simply diagonalize over the configuration space defined by
, hence
In this work the equations for
are defined in a state dependent way by projecting the Schrödinger equation (5) against excitations out of the reference state,
, where
. In practice we usually define the reference state through
, but the theory will be developed more generally, and
can in principal be obtained from a separate calculation, of MCSCF type for example. The projected equations read
The analogous equation for
would be entirely redundant however, as
is entirely contained in the
manifold of Eqn. (6). After some experimentation we found that the following is a suitable alternative
in which we essentially replaced
by the dominant part, the reference state
. The resulting equations can still exhibit exact redundancies or near singularities. This implies that quite different values for the parameters can generate (nearly) the same wave function, and it typically means that the non-linear equations are very hard to solve numerically. Similar issues arise in internally contracted MR-CI [
41] and in practice we eliminate the near redundancies, by projection on a regularized subspace. This is non-trivial and requires further discussion (see below). The above equations are coupled and we solve for the
,
amplitudes from
one iterative procedure. This means that each state has its associated set of
amplitudes and the procedure is completely state-specific. In addition we have the flexibility to use the
amplitudes (and
t1-equation) to define Brueckner orbitals.
We will also consider an alternative and simplified scheme where we define the
t2-equations as
in analogy to the equation for the
t1 coefficients.
The equations in which we use the complete operator in the t2-equations (Eqn. 7) define the Complete State Selective EOMCC approach (CSS-EOMCC), while we use the acronym RSS-EOMCC if instead of we use (Eqn. 9). As a mnemonic the acronyms contain the respective operators or , while in addition the abreviation RSS-EOMCC stands for "Restricted (or Reference) State Selective EOMCC". While in the CSS-EOMCC approach , in the RSS-EOMCC approach we take an additional short-cut by obtaining from a diagonalization of the bare Hamiltonian over the 2h configurations only (DIP-TDA), and hence is not relaxed in the presence of dynamical correlation effects. Both of these methods are initial trial versions and in this paper we simply want to investigate the sensitivity of the results to such minor variations on the theme. There are a number of details that require further discussion. Let me briefly mention the salient points and defer the full details to a future exposition. In due time I hope to establish a single robust approach in which these details are specified once and for all. It does not serve much of a purpose to invent a large set of slightly different variants except in the testing stage.
Spin-adaptation.
In the above formulation we used spin-orbitals to facilitate the discussion somewhat but this is not what is done in practice. In reality we parameterize the
operator using generators of the unitary group
and the projections used in the
T-equations are against
and
. The parameterization of the
operators in terms of the generators of the unitary group ensures that the transformed Hamiltonian
commutes with the spin-operators and the resulting wave functions are rigorous spin eigenfunctions. Let us note that the spin-adapted variant of the
equations are
not equivalent to the spin-orbital equations in this case. These issues have been discussed in detail in a previous paper of us in which we explore spin-adaptation in a CC framework [
42]. The final diagonalization of
over the
2h and
3h1p configurations is carried out in a spin orbital basis but the diagonalization manifold is easily seen to be spin adapted. In principle we could use an explicitly spin-adapted formulation in this step, and spin-orbitals are used purely for our convenience.
Elimination of near-singularities.
The reference state defines a symmetric and positive definite metric matrix
and the doubles analog
, neither of which depend on the virtual labels
a,
b. These metric matrices have orthonormal eigenvectors
Uλ and eigenvalues
λ, some of which may be small (smaller than 0.01 say). Defining projectors on the regularized subspaces
we solve the regularized equations
with similar expressions for the singles equations. The reference state entering the projectors is determined at the beginning of the iterative procedure and is kept fixed. This improves convergence, compared to projections that are determined iteratively. We have an input threshold variable to decide which eigenvalues of the metric matrix are kept in the projection, and this is set to 0.01 in the current calculations.
Size consistency, size-extensivity and size-intensivity.
If a compound system consists of an open-shell system A and an additional non-interacting closed-shell molecule B (located at infinity), there exist solutions to all of the equations in this paper that correspond exactly to the subsystems in isolation. In particular, if the molecular orbitals are taken to be localized all operators , , become completely localized on the individual subsystems. The localized equations for closed-shell subsystem B in the compound system reduce to the standard CCSD equations for system B alone. Likewise, the localized equations for open-shell subsystem A are unaffected by the presence of non-interacting subsystem B. In this case of a compound system consisting of an open-shell and a closed-shell system the orbitals can always be localized and this facilitates the analysis. However the theory is invariant under such localization and the conclusion is completely general. This is a very desirable scalability property that we refer to as size-intensivity. It presumably implies that the energies, in particular energy differences between different open-shell states, are size-extensive (= physically well-behaved) as the size of the system grows, although a precise mathematical definition of this property for an open-shell system is hard to give. Let me emphasize this point here. To my knowledge there is no theoretical framework to analyze or even define the property of size-extensivity in the thermodynamical sense for an open-shell system, because there is no repeating unit that leads to a constant energy per unit cell in the infinite limit.
While the SS-EOMCC approach separates properly into an open-shell plus closed-shell fragements (size-intensivity) it is certainly not size-consistent in the sense that if a triplet molecule separates into two doublet radicals, the energy we would calculate using the present method is not precisely twice the energy of the doublet states in isolation (calculated by the analogous method for doublet states [
43]). This is due to the linear CI diagonalization for the target states. The origin of the problem is the same as the issue with charge-transfer separability in EE-EOMCC calculations as analyzed in detail in earlier papers [
30,
44,
45].
Automated Implementation.
The details of the equations (in particular the equations for the
t2 manifold) are quite involved. When we started this work we were quite aware that we needed to be able to explore various alternatives for a possible MRCC theory. Following earlier work in this direction, in particular by Janssen and Schaefer [
46] and Li and Paldus [
47] we built a tool to aid the derivation and implementation of many-body methodology using automation. The Automatic Program Generator (APG) we developed, generates an efficient Fortran procedure to perform the calculation starting from simple input equations like the ones used in this paper. Some details can be found in our earlier paper on a MRCC theory for doublet states [
43]. The APG derives the detailed equations using essentially Wick's theorem and some further embellishments (e.g. taking symbolic derivatives, multiplying by density matrices, spin-integration). The resulting equations consist of a large number of terms (on the order of 300 spin-adapted Goldstone diagrams), consisting of multiplications of typically 3 or 4 operators. In the next step the equations are factorized so that each elementary step in the factual calculation consists of addition or contraction of two matrices. Optimal factorization constitutes an n-p complete problem in mathematics, and at present we appear to have developed a reasonable strategy to tackle the factorization issue. Finally the APG generates a Fortran subroutine that calculates the residuals of Eqns. (7-9) given a set of input amplitudes, the relevant density matrices of the reference state, and Hamiltonian matrix elements. The generated Fortran subroutines use a library of handwritten subroutines that are computationally efficient. This efficiency is illustrated by the fact that the same subroutines have been used in STEOM-CCSD calculations on free base porphyrin in a DZP+diffuse basis set [
48]. The APG is well tested [
49,
50] and can be expected to provide fairly efficient and error-free implementations of highly advanced electronic structure methods.
An additional factorization step.
In order to facilitate the computation, in particular in connection to the APG we introduce an additional approximation in the case of a CSS-EOM-CCSD calculation. In practice we factorize the
operator as
, where
, where
m indicates an active occupied orbital label. The operator
has the same form as the IP-operator in STEOM-CCSD theory (e.g. [
30]). This factorized form of
only occurs in the evaluation of the
t2 equation (Eqn. (7)), not in the final diagonalization. The reason to use this factorization step is that we can then avoid the use of three-particle intermediates in any of the factorized equations, and we can use unitary group operators throughout. Let me emphasize here that this step has a purely computational origin, and it is essential to employ this factorization in conjunction with the present version of the APG. We do introduce an active space in this step, but this is only done to fit the true
operator. The factorization is a computational device, rather than a theoretical construction, and if the CSS-EOMCC scheme were to turn out our final method of choice we can eliminate this factorization step with some additional work. In practice we solve a regularized form of the following linear equation
to obtain the coefficients of the
operator. From our results below we find that the contribution due to
itself only marginally affects the
t2 amplitudes, and hence the slight error we make in the above factorization is presumably of little importance. The choice of active space is a purely computational matter, and we simple choose it to optimize the fit. It has nothing to do with the multiconfigurational problem.
Truncation of singles.
In practice we do not fully develop the quartic expansion of the single excitation coefficients, but instead only keep up to linear terms. The reason for this is again purely technical. If Brueckner orbitals are used the results are unaffected by the truncation. For the examples considered in this work the truncation makes very little difference, as the t1-amplitudes are always small (0.02 or less).
State averaged Calculations.
It can be advantageous to perform calculations in a state averaged way. This is true in particular if degeneracies are involved (e.g. Π and Δ states for the diatomics below). The state selective determination of the
t-amplitudes can break symmetry, so that diagonalization of
would not lead to equal energies for the Π
x and Π
y states in a diatomic for example. In a state averaged calculation only the equations for the
t-amplitudes are modified:
In principal we have a flexibility to choose the weights
wλ in the sum over included states, although in practice we always keep them equal (essential to keep degeneracies). The implementation of the programs is flexible and the subroutines written by the APG take the state-averaged density matrices on input.
Brueckner MCSCF
All of the implementations were incorporated in a local version of the ACES II package [
51], which does not have any MCSCF capabilities at present. We used the APG to implement a limited MCSCF procedure through a CC singles variant of our SS-EOMCC equations. In the Brueckner MCSCF scheme we solve the following set of coupled equations
where the diagonalization of
is over the reference
2h configurations. The
t1 amplitudes are used to define a rotation of the orbitals defining
and the usual Brueckner scheme is used to iterate to convergence
.
Iterative Sequence of the Solution Algorithm
In the first step of the algorithm we find a suitable set of starting orbitals to define
. Typically we use UHF or ROHF orbitals for the triplet state, from which we construct a spatial orbital density matrix. The strongly occupied natural orbitals of this density matrix define the closed-shell reference state. This scheme is analogous to Pulay's UNO-CAS scheme [
52], and the orbitals can be further refined using the Brueckner-MCSCF procedure. We can also start from converged RHF orbitals for the dianion. After the determination of the vacuum state the algorithm has two branches depending on whether we use the CSS-EOMCC or RSS-EOMCC variant.
Steps in RSS-EOMCC calculation:
Calculate , .
Solve the RSS-EOMCC equations for
t1 and
t2 amplitudes, using DIIS [
53] to accelerate convergence.
Calculate appropriate matrix elements of (in particular the numerous and elements are not needed).
Diagonalize over the 2h and 3h1p configurations. We can easily find multiple states in this scheme. The operator is defined in a state selective fashion, but if the t-amplitudes are assumed to be transferable, a number of states can be calculated simultaneously. This latter scheme is referred to as t(ransfer)-RSS-EOMCC.
Steps in a CSS-EOMCC calculation
Calculate , .
Iterate: [ Calculate appropriate elements of
; Diagonalize over
2h and
3h1p configurations to obtain
; Fit
to
; Calculate residuals in the
and
equations and update the
t-amplitudes, using
]. In our DIIS extrapolation we use both the
and
operators (and orbital rotation parameters in a Brueckner calculation following reference [
54]).
In a final diagonalization of we can include more states and in this way gain insight into the transferability of t-amplitudes. Such calculations are acronymed t-CSS-EOMCC.
The amount of work that goes into a CSS-EOMCC calculation is substantially greater than in a RSS-EOMCC calculation: In each CSS-EOMCC iteration we form
and diagonalize over the
2h/3h1p determinants. Moreover the CSS-EOMCC
t2-equation is far more involved due to the presence of
(in the guise of
).
IV. Conclusions
In this paper we have presented results for a few state selective variants of equation of motion coupled cluster theory (SS-EOMCC). In principle all parameters: orbitals, "closed-shell" cluster coefficients and wave function amplitudes of the transformed Hamiltonian, can be optimized explicitly for the state of interest. We limited the discussion here to situations were the dominant part of the wave function can be described by deleting two electrons from a closed-shell vacuum determinant, the so-called DIP scheme. The scheme can be readily generalized to the deletion of n-electrons from an (N+n)-electron type closed-shell vacuum state, and this appears to be a promising step towards a completely general internally contracted multireference CC method. For such a general scheme it is clear that a state selective scheme is required as the vacuum state loses all physical significance. We discussed two principal variants of the DIP based SS-EOMCC scheme. In the Complete SS-EOMCC scheme the t2 amplitudes are obtained by projecting the parameterized Schrödinger equation onto a suitable excitation manifold, while in the Reference SS-EOMCC scheme we use only the dominant 2-hole part of state and project , where is obtained from a DIP-TDA calculation. The latter scheme is quite a bit simpler and it provides equally good, possibly better results than the complete variant, in particular for the absolute total energy. Both methods overall perform quite satisfactorily for the diatomic molecules considered, especially for energy differences, and it is premature to make a definitive assessment or comparison between the two approaches. More results and benchmarks need to be obtained. However, if the difference between the two approaches is consistently minor the RCC-EOMCC method is definitely to be preferred, because of its relative simplicity. Let us emphasize here that at present we do not have a rigorous formal argument to choose one approach over the other, as the amplitudes are abitrary in the limit of a complete expansion. We also investigated if the t-amplitudes that are obtained in this scheme are transferable from one state to the next, and this appears not to be the case. This is a slightly surprising result as the t-amplitudes are supposed to account purely for dynamical correlation, which is expected to be fairly similar between related states. The observations made in this study may guide us in further explorations in our quest for completely general multireference CC methods.

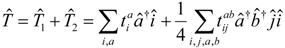
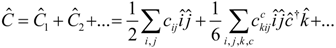
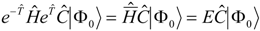



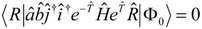
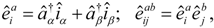
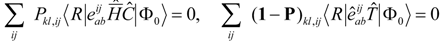
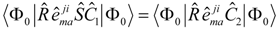
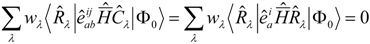
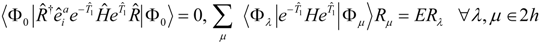
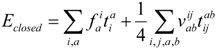 , and the total energy. For comparison in table VII we listed results for Brueckner based CSS-EOM-CCSD and RSS-EOM-CCSD evaluated at identical geometries. First of all we note that even though the formal reference determinant is the same for all states, there are significant differences in vacuum energy that are due to orbital relaxation effects. So even though all states in dioxygen have the same spatial configuration, this does not mean that the optimum spatial orbitals are the same for all states. The ground states in both O2 and F2 have the highest vacuum state energy (corresponding to the dianion), and this reflects the relative compactness of the ground state LUMO. The closed-shell parts of the correlation energy tend to be fairly similar, a clear exception being the closed-shell part of the correlation energy of the ground state for F2. There are significant differences however between the CSS and RSS variants, in particular the difference in vacuum state energy corresponding to the respective Brueckner determinants. The RSS-EOM-CCSD method universally yields lower total energies than CSS-EOM-CCSD, on the order of 10 mHartree (0.3 eV). The relative energies between various excited states are far more accurate than 0.3 eV, but it does remain a little puzzling where the discrepancy in total energy comes from. Part of it seems to be related to the vacuum energy part which can easily be different by 25 mHartree. It may be good to reiterate that in the RSS approach the reference state is obtained from a DIP-TDA calculation, diagonalizing the bare Hamiltonian over the 2-hole configurations, while subsequently the t-amplitudes are solved and the vacuum determinant is defined in a Brueckner sense. In contrast in the CSS-EOMCC approach at each step the DIP-EOMCC equations are solved and the reference state is defined as the 2-hole component of the full DIP-EOM eigenvector. So in the CSS-EOMCC approach the reference state is fully relaxed with respect to dynamical correlation. It is counterintuitive that the RSS-EOMCC approach would yield the lowest energy and this is certainly a clear manifestation of the lack of a variational principle associated with these approaches. The comparison also makes clear that to obtain accurate energy differences we are relying on a balance that leads to a cancellation of errors. I have little insight in the systematics of this balancing act at present and would certainly have preferred to get more consistent total energies.
, and the total energy. For comparison in table VII we listed results for Brueckner based CSS-EOM-CCSD and RSS-EOM-CCSD evaluated at identical geometries. First of all we note that even though the formal reference determinant is the same for all states, there are significant differences in vacuum energy that are due to orbital relaxation effects. So even though all states in dioxygen have the same spatial configuration, this does not mean that the optimum spatial orbitals are the same for all states. The ground states in both O2 and F2 have the highest vacuum state energy (corresponding to the dianion), and this reflects the relative compactness of the ground state LUMO. The closed-shell parts of the correlation energy tend to be fairly similar, a clear exception being the closed-shell part of the correlation energy of the ground state for F2. There are significant differences however between the CSS and RSS variants, in particular the difference in vacuum state energy corresponding to the respective Brueckner determinants. The RSS-EOM-CCSD method universally yields lower total energies than CSS-EOM-CCSD, on the order of 10 mHartree (0.3 eV). The relative energies between various excited states are far more accurate than 0.3 eV, but it does remain a little puzzling where the discrepancy in total energy comes from. Part of it seems to be related to the vacuum energy part which can easily be different by 25 mHartree. It may be good to reiterate that in the RSS approach the reference state is obtained from a DIP-TDA calculation, diagonalizing the bare Hamiltonian over the 2-hole configurations, while subsequently the t-amplitudes are solved and the vacuum determinant is defined in a Brueckner sense. In contrast in the CSS-EOMCC approach at each step the DIP-EOMCC equations are solved and the reference state is defined as the 2-hole component of the full DIP-EOM eigenvector. So in the CSS-EOMCC approach the reference state is fully relaxed with respect to dynamical correlation. It is counterintuitive that the RSS-EOMCC approach would yield the lowest energy and this is certainly a clear manifestation of the lack of a variational principle associated with these approaches. The comparison also makes clear that to obtain accurate energy differences we are relying on a balance that leads to a cancellation of errors. I have little insight in the systematics of this balancing act at present and would certainly have preferred to get more consistent total energies.