1. Introduction
The term “mixed electronic states” refers to states in which electronic structures of different types, such as valence and Rydberg, or covalent and ionic, contribute strongly to the wave function. Moreover, the relative contributions of the different types tend to vary strongly with variations in the molecular geometry, as in the case of avoided curve crossings. In multireference electronic structure calculations for such states, the relative contributions of different reference configurations tend to differ substantially between the reference (MCSCF) wave function and the final correlated wave function. For example, the crossing point between the potential energy curves of the covalent and ionic configuration in alkali halides can vary by several Bohr between an MCSCF and a multireference CI calculation [
1] (primarily because of the great difficulty in reproducing the electron affinity of the halogen atom). Obviously, in the region between the MCSCF and correlated crossing points, the MCSCF solution provides the wrong zero-order functions for the multireference treatment. If the computational model does not allow relaxation of the coefficients of the reference configurations in the correlation treatment, a correct description of the mixing and of the potential energy surfaces cannot be expected.
Two examples of correlation treatments that are affected by the problem of incorrect mixing are internally-contracted CI [
2,
3,
4,
5] and state-specific multireference perturbation expansions (of the diagonalize-then-perturb variety) at second and third order, such as CASPT2 [
6] and CASPT3 [
7]. The reference (MCSCF) function does not interact directly with its orthogonal complement in the reference space, and therefore the other eigenvectors of the MCSCF Hamiltonian do not contribute to a perturbation expansion before the fourth order in the energy. Solutions for this problem in the case of contracted CI have been introduced [
5,
8], in which contracted CI excitations based on more than one MCSCF reference-space eigenvector are included. Similar ideas have been applied in multireference perturbation theory, as discussed below, and one form of such an approach for second- and third-order multireference Rayleigh–Schrödinger perturbation theory is described here.
Multireference perturbation approaches fall into two main classes: In quasidegenerate perturbation theory [
9,
10,
11], also referred to as “perturb then diagonalize,” a low-dimensional effective Hamiltonian is constructed using a perturbation expansion for each of its matrix elements, and this effective Hamiltonian is diagonalized to obtain the desired solutions for one or more states. The principal difficulty with this approach is the problem of intruder states, which can cause the perturbation series to diverge or to converge extremely slowly. On the other hand, in state-specific perturbation theory, also called “diagonalize then perturb,” a zero-order function is first constructed by diagonalizing the Hamiltonian over the reference space, usually by an MCSCF calculation, and a single perturbation expansion is then constructed over this zero-order function. The principal problem with this approach is the previously mentioned lack of relaxation of the zero-order function before reaching the computationally demanding fourth order in the energy expansion.
The present paper discusses a form of multireference perturbation theory that can be referred to as a “diagonalize-then-perturb-then-diagonalize” approach, which is designed to facilitate the relaxation of the reference function and thus deal with the mixed-states problem. It begins with a state-averaged MCSCF [
12,
13] calculation to provide a small number of model-state zero-order functions, and applies quasidegenerate perturbation theory to obtain an effective Hamiltonian in that small model space, followed by diagonalization of the effective Hamiltonian to obtain properly-mixed wave functions and energies. The model states used to construct the effective Hamiltonian are a small subset of the eigenstates of the state-averaged MCSCF Hamiltonian, and if they can be chosen to be well-separated in energy from any other zero-order states the intruder-state problem can be reduced.
This approach has been proposed and implemented, in one form or another, by a number of researchers, including Malrieu and co-workers [
14], Sheppard et al. [
15], Lisini and Decleva [
16], Nakano [
17], and Roos and co-workers [
18]. The current presentation includes the following specific features:
The choices of orbitals and zero-order Hamiltonian try to mimic the Møller–Plesset procedures that have been found to be very effective in single-reference perturbation expansions.
The zero-order Hamiltonians need not be diagonal.
A non-Hermitian effective Hamiltonian is generated, based on the Bloch equation.
The method is formulated in configuration space, allowing flexibility in the choice of the reference space (including incomplete active spaces), and enabling easy implementation in a CI program.
Uncontracted configuration state functions are used as a basis for the perturbation expansions.
Procedures for both second and third order in the energy are included.
The other presentations share many of these features and differ in various respects, such as in the use of natural orbitals, Epstein-Nesbet partitioning, complete active spaces, diagonal zero-order Hamiltonians, Hermitian effective Hamiltonians, many-body methods, or limitation to second-order energies.
In order to introduce the notation and the overall approach, the fundamentals of Rayleigh–Schrödinger perturbation theory are reviewed very briefly in a general form in
Section 2, and the application to the state-specific multireference treatment is described in
Section 3. The generalization for mixed states is described in
Section 4, followed by discussion in
Section 5.
2. Rayleigh–Schrödinger perturbation theory for arbitrary zero-order functions
Conventionally, a perturbation treatment begins with the partitioning of the Hamiltonian,
into a zero-order part
, for which the solutions are known, and a perturbation
. Then the desired solutions are expanded, order by order, in the eigenfunctions of
. Here we shall use the alternative, more general approach, in which the process is reversed, beginning with a given set of expansion functions and defining
in terms of these functions. This approach clearly demonstrates the various options available in the method, and is often followed in electronic structure presentations.
Given an orthonormal set of zero-order functions
of which one, labeled
, is an approximation for the state of interest, we define a zero-order Hamiltonian
with the Hermitian matrix
E(0) defined so that
and
. We then have
In principle, the choice of the
is arbitrary; however, an appropriate choice is essential, since it determines the convergence rate of the perturbation series. The perturbation is given by
and the perturbation series for the state approximated by
is
We also define
The first-order energy is given in terms of the zero-order wave function,
The equation for the first-order correction to the wave function is
Expanding the first-order wave function in the zero-order functions,
and applying
from the left, the first-order equation becomes
resulting in a linear system of equations for the
:
where
. Intermediate normalization is imposed by the choice
.
In many cases it is convenient to choose an
that is diagonal in the zero-order functions, putting
, so that
in which case the linear system has the explicit solution
In either case, the second- and third-order energy corrections are given by
4. The treatment of mixed electronic states
Mixed electronic states are characterized by large changes in the relative contributions of the reference configurations in a well-correlated final wave function compared to the zero-order MCSCF function. Typically the reason for this behavior is that two or more zero-order wave functions are close in energy and can mix strongly in the wave functions for the corresponding correlated electronic states. Examples are provided by avoided crossing regions in potential energy curves and surfaces and by states that involve mixing of valence and Rydberg character. The number of zero-order functions involved in the mixing is usually quite small, typically just a few multiconfigurational functions.
This problem can be dealt with by the use of quasidegenerate perturbation theory (QDPT) based on a model space consisting of the few zero-order functions expected to mix strongly in the states of interest. These zero-order functions (“model states”) are a small subset of the multiconfigurational expansions obtained in the diagonalization of an MCSCF Hamiltonian over the reference space. An effective Hamiltonian is then constructed in second order and, if desired, third order and diagonalized to provide improved energies and wave functions. While the reference space may be quite large, only a few model states (each of which is an expansion over all the reference configurations) are used, and if these model states can be chosen to be well separated in energy from other states, the intruder-state problem common in QDPT treatments is not likely to be significant in this approach. However, it may be difficult to maintain such a separation while avoiding discontinuities in the model space over a wide range of geometries on a potential energy surface.
The procedure begins with a state-averaged MCSCF calculation in order to determine orbitals that represent a compromise between those most suitable for each of the individual zero-order functions likely to be involved in the mixed states. The coefficients of the reference configurations obtained in the MCSCF diagonalization for the contributing states provide a number of zero-order vectors (α = 1, 2, …, l, l << m) defining the corresponding model-state zero-order functions . These functions are a small subset of the functions , β = 1, 2, …, m, discussed in the previous section, and like them, are orthonormal and noninteracting over (and over ). The vectors are collected into a matrix C(0) of l columns, in which at most the first m rows (corresponding to the reference configurations) are nonzero.
The zero-order Hamiltonian is chosen as in Eq. (19), with the whole reference block of
E(0) taken to be diagonal, so that we have
and elements of
E(0) involving orthogonal-complement functions
,
l <
β ≤
m, do not enter into the calculation of the first-order wave function and the second- and third-order energies.
The effective Hamiltonian
is expanded as
where
is known as the
shift operator. Only the model-space part of these operators is needed, and can be represented by
l ×
l matrices,
where the zero-order part is simply the diagonal model-space block of
E(0),
We also define
The first-order effective Hamiltonian is simply the corresponding portion of the diagonalized MCSCF Hamiltonian,
The effective Hamiltonian is related to the original Hamiltonian by a similarity transformation that decouples the model-space part from the rest of the Hilbert space,
where the
l-column decoupling matrix
C, which is a representation of the
wave operator, has the order-by-order expansion
in terms of the coefficient matrices defining the order-by-order expansions of the wave functions. As before, the matrix
H (unlike
H0 in Eq. (38)) is defined in terms of the individual configuration state functions Θ
i. Equation (41) is a representation of the Bloch equation [
9] in the configuration-state-function basis. Note that the first
m rows of
ΔC(1) are zero because of intermediate normalization and the noninteracting nature of the functions
(
α ≤
m).
The first-order wave operator is represented by the
l-column matrix
C(1) =
C(0) +
ΔC(1) constructed from the first-order vectors
(
α = 1, 2, …,
l) calculated for each of the zero-order functions by Eq. (12) or (14), analogously to the single-state calculations described in the previous section. The second-order non-Hermitian shift operator is then obtained as
Solution of the
l ×
l non-Hermitian eigenvalue problem
provides the second-order energies in the diagonal matrix of eigenvalues
![Ijms 03 00639 i033]()
and the expansion coefficients of the properly-mixed first-order wave functions
by the transformation
For the calculation of the third-order shift operator we note that Wigner's (2
n + 1) rule does not apply to the off-diagonal elements of the non-Hermitian shift operator, and therefore
W(3) cannot be fully obtained from the first-order wave functions. Instead it is computed from the second-order wave functions,
The complete second-order wave functions include contributions from triple and quadruple excitations and from the orthogonal complement space {
(
l <
γ ≤
m)}. However, as seen from Eq. (47), only those terms that interact (across
) with the zero-order functions, i.e., the single- and double-excitation terms, are required. If we choose
E(0) to have no off-diagonal elements coupling the higher excitations with the single and double excitations, we can decouple the equations for the single- and double-excitation coefficients from the others and solve for these from the linear equations system
In fact, in the diagonal case these coefficients are obtained directly from
In either case, the principal computational step is the calculation of the matrix-vector products
so that the computational effort is not very different from
l times the work required for the calculation of the third-order energy for the single state case, Eq. (34).
The third-order shift-operator matrix elements are easily obtained by the contraction of vector pairs,
This step is followed by the solution of the non-Hermitian eigenvalue problem for the third-order effective Hamiltonian matrix
H(3) to obtain the third-order energies
![Ijms 03 00639 i041]()
. While a transformation matrix
X(2) is also obtained, and can be used to obtain the transformed coefficients matrix
for the relevant part of the second-order wave functions
, it does not provide the complete second-order wave function, because of the omission of the higher excitations and the orthogonal complement functions. Once the truncated second-order wave function has been obtained, the construction of the third-order effective Hamiltonian matrix and the diagonalization require little computational effort, and thus the total effort is still about
l times the effort in the single-state case.
An important aspect in which this treatment differs from the single-state case is the fact that it is no longer possible to tailor the zero-order Hamiltonian to resemble closely an appropriate Fock operator for each of the states in question, because of the use of state-averaged MCSCF. This aspect is the most serious shortcoming of the approach, especially in cases in which the model states differ greatly in the character of their orbitals. Furthermore, the most reasonable choices for the generalized Fock operator, and thus for , are likely to involve more nonzero off-diagonal elements in E(0) and V, and to be less analogous to the Møller–Plesset partitioning of single-reference perturbation theory. Nevertheless, the E(0) matrix should remain very sparse, because it is the matrix representation of a one-electron operator (the state-averaged Fock operator). Another potential problem is that it may be very difficult to choose a model space that does not vary discontinuously over a potential energy surface.
5. Discussion
The principal advantage of Rayleigh–Schrödinger perturbation theory over configuration interaction is the
extensivity of the energy in each order of perturbation, i.e., the proper scaling of the energy with the extent of the system [
28]. This property is a consequence of the true order-by-order nature of the Rayleigh–Schrödinger perturbation expansion (unlike Brillouin–Wigner perturbation theory, in which the infinite-order energy appears in each order of the energy expression). In a many-body formulation extensivity can be determined by demonstrating the absence of unlinked-diagram terms in the energy expressions. However, multireference many-body treatments using incomplete model spaces generally contain some unlinked diagrams, and thus are not exactly extensive [
29,
30]. In the present, configuration-based treatment the diagrammatic analysis is not easily applied. As in the many-body approach, it is to be expected that extensivity of the proposed method would depend on the choice of the model space, but the true order-by-order nature of the expansion should help in providing approximate extensivity .
A related property is
size consistency [
31] (or
strict separability [
26]), which requires that the energy calculated for a system consisting of noninteracting fragments be equal to the sum of the energies, calculated by the same model, for the separate fragments. This property ensures proper description of bond breaking. However, a perturbation expansion cannot be size consistent, in general, unless the reference function is size consistent. In a Hartree–Fock-based single-reference perturbation expansion (Møller–Plesset perturbation theory) size consistency usually requires the use of an unrestricted Hartree–Fock (UHF) reference function, which has undesirable consequences [
32,
33]. In multireference methods size consistency is not easily defined because of the difficulty in specifying equivalent reference spaces for the total system and its fragments. Therefore, the approach normally used in describing bond breaking and other processes in multireference calculations is to treat the system as a single unit in all its configurations, including its dissociation asymptotes. This procedure, called the
supermolecule approach, usually produces satisfactory dissociation energies even in truncated CI calculations [
34] (which are not size consistent by the usual definition), provided the reference function dissociates properly. In a single-state expansion based on an MCSCF zero-order function the reference space can be chosen to ensure proper dissociation of that function and, using the supermolecule approach, of the order-by-order energy results (assuming there are no other approximations, such as omission of terms from the summations). It is likely that the same statement can be applied to the procedure discussed here for mixed states.
There have been many formulations of multireference perturbation theory, both state-specific and quasidegenerate [
6,
7,
21,
22,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44,
45,
46,
47,
48,
49,
50,
51,
52]. Several (e.g., [
21,
22,
52]) have focused specifically on achieving extensivity and/or size consistency (see also [
53,
54]). The single-state treatment described in
Section 3 is similar to the approach of Hirao [
48], differing from it primarily in how the flexibility in the choice of the MCSCF orbitals is used and, therefore, in the choice of the Fock matrix elements. Unlike CASPT2 [
6] and CASPT3 [
7], the present treatment does not assume a complete-active-space reference space. On the other hand, the CASPT methods expand the perturbed wave functions in contracted excited functions, rather than in the individual configuration state functions used in the present approach, resulting in much shorter expansions (at the cost of a more complicated procedure). The multi-state generalization shares some ideas with extensions to the contracted CI approach for dealing with mixed stated [
8]. The key elements in the approach described here are the attempt to mimic Møller–Plesset partitioning and the use of just a few state-averaged MCSCF wave functions to define the model space for a QDPT calculation of the second- and third-order energies. The method treats the several mixing states symmetrically, and since it is configuration based rather than a many-body approach, it avoids the difficulties that the use of such model states would present for a diagrammatic formalism. However, this gain is obtained at the probable cost of a less efficient procedure than may be possible in a diagrammatic method.
A diagonal version of the single-state approach described in
Section 3 was reported by Shavitt and Stahlberg in 1991 [
55,
56] after implementation and testing in the
Columbus program system [
57]. This implementation was later generalized to the nondiagonal case [
57(b)]. The multistate formalism has been implemented in a development version of the
Columbus program system. It differs from the method of Roos and co-workers [
18] in several respects: It is not limited to complete-active-space reference spaces; it expands the perturbed wave functions in uncontracted excited configuration state functions instead of contracted excitation functions; and it includes the third-order capability. Test applications of this formalism have reproduced the second-order results of Roos and co-workers [
18(b)] for the avoided crossing in LiF and for the V-state of ethylene, and produced moderate improvements in third order [
58].
The idea of a model space focusing on just a few model states, much smaller in number than the size of the reference space in which the model states are constructed, is also a feature of the intermediate Hamiltonian method of Malrieu and co-workers [
59]. Among reviews of multireference perturbation methods are those by Malrieu and co-workers [
59] and Hirao and co-workers [
60].


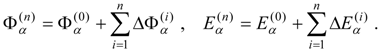


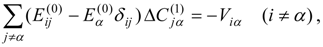
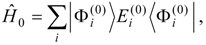
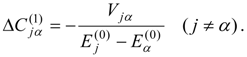
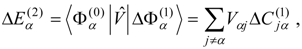
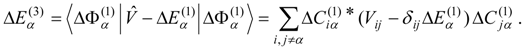
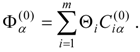

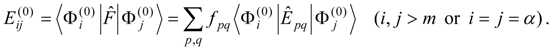
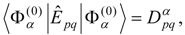
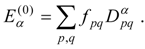
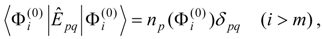
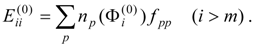
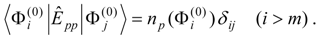
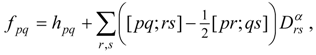
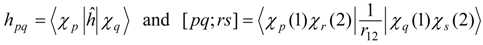
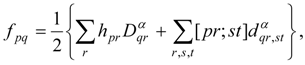
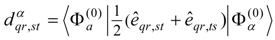
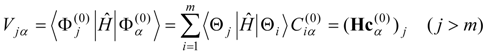
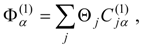
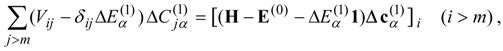
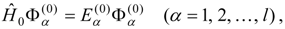
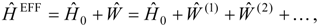
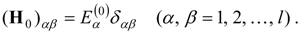
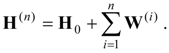
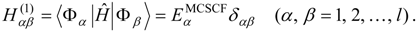
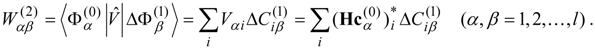
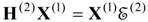
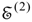 and the expansion coefficients of the properly-mixed first-order wave functions
and the expansion coefficients of the properly-mixed first-order wave functions
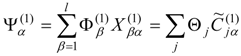

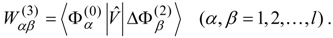

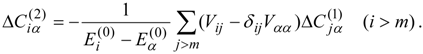
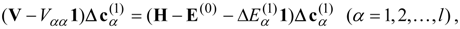
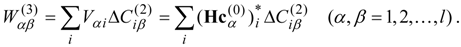
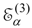 . While a transformation matrix X(2) is also obtained, and can be used to obtain the transformed coefficients matrix for the relevant part of the second-order wave functions , it does not provide the complete second-order wave function, because of the omission of the higher excitations and the orthogonal complement functions. Once the truncated second-order wave function has been obtained, the construction of the third-order effective Hamiltonian matrix and the diagonalization require little computational effort, and thus the total effort is still about l times the effort in the single-state case.
. While a transformation matrix X(2) is also obtained, and can be used to obtain the transformed coefficients matrix for the relevant part of the second-order wave functions , it does not provide the complete second-order wave function, because of the omission of the higher excitations and the orthogonal complement functions. Once the truncated second-order wave function has been obtained, the construction of the third-order effective Hamiltonian matrix and the diagonalization require little computational effort, and thus the total effort is still about l times the effort in the single-state case.