The State-Universal Multi-Reference Coupled-Cluster Theory: An Overview of Some Recent Advances

Abstract
:1 Introduction
2 The State-Universal Multi-Reference Coupled-Cluster Theory
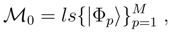

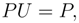
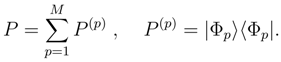
 , so that
, so that
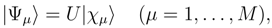
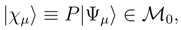
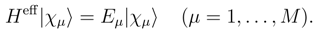
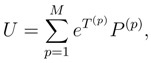 , when acting on the corresponding |Φp〉. The wave operator U, Eq. (12), reduces to the well-known wave operator of the single-reference CC theory, USRCC = eT |Φ〉〈Φ|, when model space 0 is a one-dimensional space spanned by a single reference configuration |Φ〉.
, when acting on the corresponding |Φp〉. The wave operator U, Eq. (12), reduces to the well-known wave operator of the single-reference CC theory, USRCC = eT |Φ〉〈Φ|, when model space 0 is a one-dimensional space spanned by a single reference configuration |Φ〉.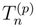 . In the exact SUMRCC formalism,
. In the exact SUMRCC formalism,
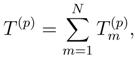

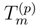 is obtained by replacing Eq. (7) by the equation HU|Φp〉 = UHU|Φp〉, where U is defined by Eq. (12), premultiplying this equation on the left by e−T(p), and projecting the resulting equation on the excited configurations relative to |Φp〉 belonging to . The final equations for cluster operators defining the exact SUMRCC theory can be written as follows:
is obtained by replacing Eq. (7) by the equation HU|Φp〉 = UHU|Φp〉, where U is defined by Eq. (12), premultiplying this equation on the left by e−T(p), and projecting the resulting equation on the excited configurations relative to |Φp〉 belonging to . The final equations for cluster operators defining the exact SUMRCC theory can be written as follows:
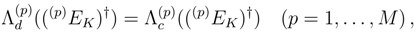
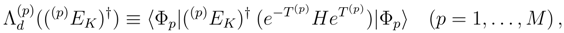
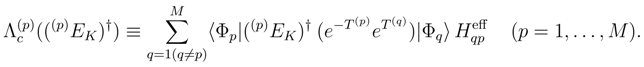 , when acting on |Φp〉. These operators are also used to represent cluster operators T(p),
, when acting on |Φp〉. These operators are also used to represent cluster operators T(p),
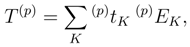
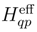 quantities entering the coupling term
quantities entering the coupling term  , Eq. (17), are the matrix elements of the SUMRCC effective Hamiltonian,
, Eq. (17), are the matrix elements of the SUMRCC effective Hamiltonian,
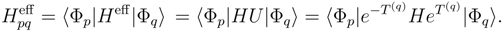
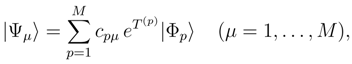
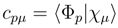
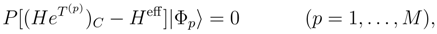
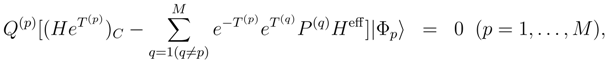
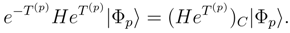 that are generated by excitation operators (p)EK. If
that are generated by excitation operators (p)EK. If  designates a projection operator onto the subspace of
designates a projection operator onto the subspace of 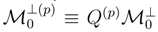 spanned by the n-tuply excited configurations relative to |Φp〉 that belong to , we can write
spanned by the n-tuply excited configurations relative to |Φp〉 that belong to , we can write
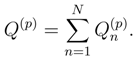
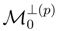 , used to define cluster operators T(p), are identical, i.e., = for all p = 1, . . . , M. This is a consequence of the fact that
, used to define cluster operators T(p), are identical, i.e., = for all p = 1, . . . , M. This is a consequence of the fact that
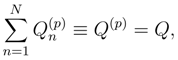 of cluster operators
of cluster operators 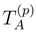 , namely,
, namely,
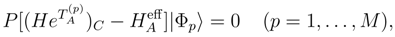
 are defined by Eq. (14),
are defined by Eq. (14),  is the effective Hamiltonian of the approximate SUMRCC method A, and
is the effective Hamiltonian of the approximate SUMRCC method A, and 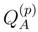 is a projection operator onto the excited configurations used to define , i.e.,
is a projection operator onto the excited configurations used to define , i.e.,
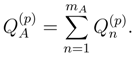
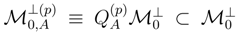 , used to define the truncated cluster operators , are usually different for different reference configurations |Φp〉 (cf., e.g., the lists of excitations (p)EK, corresponding to different references |Φp〉 of the OSA SUMRCCSD formalism of Refs. 39, 41, 45–50, 54, given in Ref. 47). As pointed out in Ref. 111, this asymmetric treatment of the manifolds of excitations corresponding to different reference configurations causes that the approximate SUMRCC schemes based on Eqs. (27) and (28) (including the existing SUMRCCSD methods) are not equivalent to any Hermitian eigenvalue problem. This significant distortion of the exact SUMRCC theory, resulting from the truncation of the many-body expansions of all operators T(p) at the same excitation level mA,leads to a number of pathologies in approximate SUMRCC calculations based on Eqs. (27) and (28). These pathologies include the existence of an excessive number of real and complex solutions that lack physical interpretation and the appearance of the intruder solution problem [57]. The multi-reference extension of the MMCC theory, which we recently suggested in Ref. 117, and which we overview in the next section, offers a possibility of reducing the severity of problems encountered in the standard SUMRCC (e.g., SUMRCCSD) calculations by incorporating higher-order effects in the SUMRCC formalism and by reinforcing the symmetric treatment of the
, used to define the truncated cluster operators , are usually different for different reference configurations |Φp〉 (cf., e.g., the lists of excitations (p)EK, corresponding to different references |Φp〉 of the OSA SUMRCCSD formalism of Refs. 39, 41, 45–50, 54, given in Ref. 47). As pointed out in Ref. 111, this asymmetric treatment of the manifolds of excitations corresponding to different reference configurations causes that the approximate SUMRCC schemes based on Eqs. (27) and (28) (including the existing SUMRCCSD methods) are not equivalent to any Hermitian eigenvalue problem. This significant distortion of the exact SUMRCC theory, resulting from the truncation of the many-body expansions of all operators T(p) at the same excitation level mA,leads to a number of pathologies in approximate SUMRCC calculations based on Eqs. (27) and (28). These pathologies include the existence of an excessive number of real and complex solutions that lack physical interpretation and the appearance of the intruder solution problem [57]. The multi-reference extension of the MMCC theory, which we recently suggested in Ref. 117, and which we overview in the next section, offers a possibility of reducing the severity of problems encountered in the standard SUMRCC (e.g., SUMRCCSD) calculations by incorporating higher-order effects in the SUMRCC formalism and by reinforcing the symmetric treatment of the 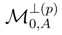 subspaces.
subspaces.3 A New Type of the Noniterative Corrections to Multi-Reference Coupled-Cluster Energies: The Method of Moments of the State-Universal Multi-Reference Coupled-Cluster Equations

 , obtained by solving the approximate SUMRCC (e.g., SUMRCCSD) equations, recover the exact (full CI) energies Eµ of the electronic states of interest. The main purpose of the approximate MM-SUMRCC calculations is to estimate corrections δµ, so that the resulting energies + δµ remain very close to the corresponding exact energies Eµ.
, obtained by solving the approximate SUMRCC (e.g., SUMRCCSD) equations, recover the exact (full CI) energies Eµ of the electronic states of interest. The main purpose of the approximate MM-SUMRCC calculations is to estimate corrections δµ, so that the resulting energies + δµ remain very close to the corresponding exact energies Eµ.
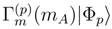 , Eq. (31), can be viewed as the most fundamental quantities of the Jeziorski-Monkhorst theory, as defined by Eqs. (27) and (28), since the system of equations for the many-body components of cluster operators , p = 1, . . . , M, Eq. (28), is immediately obtained by imposing a requirement that the lowest moments , with m = 1, . . . , mA, vanish, i.e.,
, Eq. (31), can be viewed as the most fundamental quantities of the Jeziorski-Monkhorst theory, as defined by Eqs. (27) and (28), since the system of equations for the many-body components of cluster operators , p = 1, . . . , M, Eq. (28), is immediately obtained by imposing a requirement that the lowest moments , with m = 1, . . . , mA, vanish, i.e.,
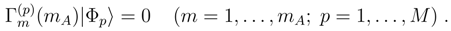 , which enter the formula for the noniterative corrections δµ, Eq. (30), are those with m > mA. As shown in Ref. 117, the explicit formula for corrections δµ, in terms of moments , is as follows:
, which enter the formula for the noniterative corrections δµ, Eq. (30), are those with m > mA. As shown in Ref. 117, the explicit formula for corrections δµ, in terms of moments , is as follows:
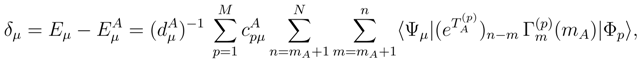
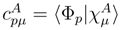 are the coefficients defining the model-space states
are the coefficients defining the model-space states
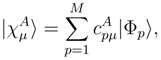
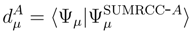
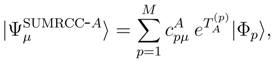
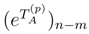 is the (n − m)-body component of
is the (n − m)-body component of 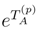 .
.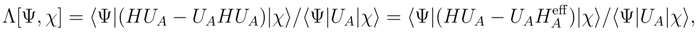
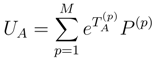
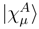 of the effective Hamiltonian , we immediately obtain
of the effective Hamiltonian , we immediately obtain
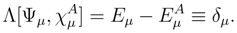
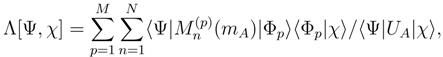

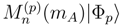 can be expressed in terms of the SUMRCC moments
can be expressed in terms of the SUMRCC moments 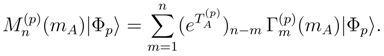 , with m = 1, . . . , mA, vanish [cf. Eq. (32)] allow us to write
, with m = 1, . . . , mA, vanish [cf. Eq. (32)] allow us to write
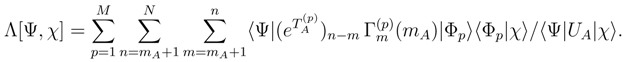 in Eq. (43) and using Eqs. (38) and (39), we obtain the desired Eq. (33)., p = 1, . . . , M, and the corresponding effective Hamiltonian, . Next, we construct the generalized moments with m > mA using Eq. (31). Finally, we use cluster operators , generalized moments , and the right eigenvectors of , i.e.,
in Eq. (43) and using Eqs. (38) and (39), we obtain the desired Eq. (33)., p = 1, . . . , M, and the corresponding effective Hamiltonian, . Next, we construct the generalized moments with m > mA using Eq. (31). Finally, we use cluster operators , generalized moments , and the right eigenvectors of , i.e., 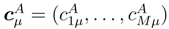 , to calculate corrections δµ with the help of Eq. (33).
, to calculate corrections δµ with the help of Eq. (33).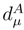 denominator term in Eq. (33) reduces to
denominator term in Eq. (33) reduces to 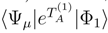 , since the model-space state becomes proportional to the reference configuration |Φ1〉. In addition, when M = 1, the generalized moments of the SUMRCC equations, Eq. (31), reduce to the generalized moments of the single-reference CC equations defined in Refs. 7, 118, 119. We obtain [cf. Eq. (31)],
, since the model-space state becomes proportional to the reference configuration |Φ1〉. In addition, when M = 1, the generalized moments of the SUMRCC equations, Eq. (31), reduce to the generalized moments of the single-reference CC equations defined in Refs. 7, 118, 119. We obtain [cf. Eq. (31)],
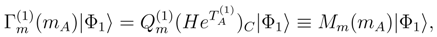
 are the generalized moments of the single-reference CC equations. The substitution of Eq. (44) into the M = 1 variant of Eq. (33) leads to the following result:
are the generalized moments of the single-reference CC equations. The substitution of Eq. (44) into the M = 1 variant of Eq. (33) leads to the following result:

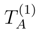 and |Φ1〉 with, respectively, the cluster operator and the reference configuration of the standard singlereference CC method., may give very good estimates of the exact energies Eµ, if we use simple estimates of wave functions |Ψµ〉, provided by one of the relatively inexpensive ab initio methods. Independent of the approximate form of |Ψµ〉 chosen for such calculations, corrections δµ can be calculated in a state-specific manner. Our belief that simple estimates of wave functions |Ψµ〉 may be sufficient to obtain accurate δµ values is based on the success of the single-reference MMCC methods and their renormalized CC analogs [7,26,117,118,119,120,121,122,123,131,132,133,134], in which simple perturbative or CI wave functions are used to construct the relevant energy corrections.
and |Φ1〉 with, respectively, the cluster operator and the reference configuration of the standard singlereference CC method., may give very good estimates of the exact energies Eµ, if we use simple estimates of wave functions |Ψµ〉, provided by one of the relatively inexpensive ab initio methods. Independent of the approximate form of |Ψµ〉 chosen for such calculations, corrections δµ can be calculated in a state-specific manner. Our belief that simple estimates of wave functions |Ψµ〉 may be sufficient to obtain accurate δµ values is based on the success of the single-reference MMCC methods and their renormalized CC analogs [7,26,117,118,119,120,121,122,123,131,132,133,134], in which simple perturbative or CI wave functions are used to construct the relevant energy corrections. 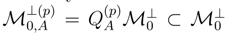 subspaces spanned by the excited configurations relative to |Φp〉 are usually different for different p values. As mentioned earlier, this asymmetric treatment of manifolds of excitations corresponding to different references |Φp〉 causes that the conventional SUMRCC approaches based on Eqs. (27) and (28) are not equivalent to any Hermitian eigenvalue problem which, in turn, leads to various problems in SUMRCC calculations. However, if we do not truncate the summations over n and m in Eq. (33) in any arbitrary manner and if we simply let the projection onto a suitably chosen approximate wave function |Ψµ〉 select terms in the summations over p, n, and m in the numerator of Eq. (33), we will obtain a fully symmetric treatment of the subspaces corresponding to different references |Φp〉. In order for this scheme to work, we only have to assume that the CI expansions of wave functions |Ψµ〉 contain some configurations whose excitation level relative to at least one of the M references |Φp〉 exceeds mA. This is certainly true for the MRCISD wave functions and their CISDt and CISDtq analogs if we are interested in correcting the SUMRCCSD results. The projection onto |Ψµ〉 in the numerator of Eq. (33) will select precisely those subsets of the generalized moments (usually, different subsets of Γ(p)’s for different values of p) that are needed to restore a symmetric treatment of the manifolds of excitations in the approximate SUMRCC (e.g., SUMRCCSD) calculations. Although this particular way of improving the SUMRCCSD results by using the MRCISD, CISDt, or CISDtq wave functions |Ψµ〉 in Eq. (33) has not been tested yet in actual numerical calculations, we believe that we should be able to obtain significant improvements in the calculated SUMRCC energies, particularly in regions plagued by intruder states or intruder solutions, where there is an apparent need to incorporate higher–than–doubly excited clusters and have a more symmetric treatment of the subspaces in the SUMRCC calculations.
subspaces spanned by the excited configurations relative to |Φp〉 are usually different for different p values. As mentioned earlier, this asymmetric treatment of manifolds of excitations corresponding to different references |Φp〉 causes that the conventional SUMRCC approaches based on Eqs. (27) and (28) are not equivalent to any Hermitian eigenvalue problem which, in turn, leads to various problems in SUMRCC calculations. However, if we do not truncate the summations over n and m in Eq. (33) in any arbitrary manner and if we simply let the projection onto a suitably chosen approximate wave function |Ψµ〉 select terms in the summations over p, n, and m in the numerator of Eq. (33), we will obtain a fully symmetric treatment of the subspaces corresponding to different references |Φp〉. In order for this scheme to work, we only have to assume that the CI expansions of wave functions |Ψµ〉 contain some configurations whose excitation level relative to at least one of the M references |Φp〉 exceeds mA. This is certainly true for the MRCISD wave functions and their CISDt and CISDtq analogs if we are interested in correcting the SUMRCCSD results. The projection onto |Ψµ〉 in the numerator of Eq. (33) will select precisely those subsets of the generalized moments (usually, different subsets of Γ(p)’s for different values of p) that are needed to restore a symmetric treatment of the manifolds of excitations in the approximate SUMRCC (e.g., SUMRCCSD) calculations. Although this particular way of improving the SUMRCCSD results by using the MRCISD, CISDt, or CISDtq wave functions |Ψµ〉 in Eq. (33) has not been tested yet in actual numerical calculations, we believe that we should be able to obtain significant improvements in the calculated SUMRCC energies, particularly in regions plagued by intruder states or intruder solutions, where there is an apparent need to incorporate higher–than–doubly excited clusters and have a more symmetric treatment of the subspaces in the SUMRCC calculations.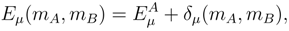
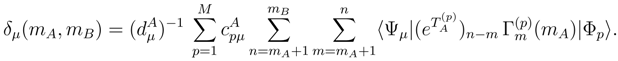 with m = mA + 1, . . . , mB. For typical applications of Eq. (46) (e.g., mA = 2 and mB = 3 or 4), the moments with m = mA + 1, . . . , mB form a small subset of all Γ(p)’s.
with m = mA + 1, . . . , mB. For typical applications of Eq. (46) (e.g., mA = 2 and mB = 3 or 4), the moments with m = mA + 1, . . . , mB form a small subset of all Γ(p)’s.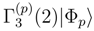 moments. The MM-SUCC(2,3) energy expression is [117]
moments. The MM-SUCC(2,3) energy expression is [117]
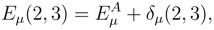
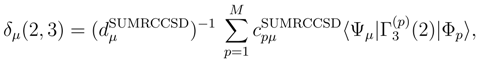
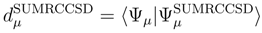
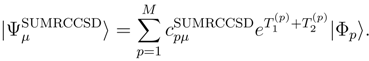
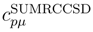 , p = 1, . . . , M, are the components of the right eigenvector of the SUMRCCSD effective Hamiltonian, whose matrix elements, designated here by (2), are defined as follows [cf. Eqs. (19) and (24)]:
, p = 1, . . . , M, are the components of the right eigenvector of the SUMRCCSD effective Hamiltonian, whose matrix elements, designated here by (2), are defined as follows [cf. Eqs. (19) and (24)]:
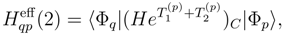
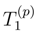 and
and 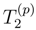 are the singly and doubly excited clusters of the SUMRCCSD approach. The moments that appear in Eq. (49) can be expressed in terms of the projections of the SUMRCCSD equations on the triply excited configurations relative to |Φp〉. If i, j, k, . . . (a, b, c, . . .) represent the spin-orbitals that are occupied (unoccupied) in the reference configuration |Φp〉 and if
are the singly and doubly excited clusters of the SUMRCCSD approach. The moments that appear in Eq. (49) can be expressed in terms of the projections of the SUMRCCSD equations on the triply excited configurations relative to |Φp〉. If i, j, k, . . . (a, b, c, . . .) represent the spin-orbitals that are occupied (unoccupied) in the reference configuration |Φp〉 and if  are the excitation operators that generate the triply excited configurations relative to |Φp〉 (Xa and Xi are the usual creation and annihilation operators, respectively), we can write
are the excitation operators that generate the triply excited configurations relative to |Φp〉 (Xa and Xi are the usual creation and annihilation operators, respectively), we can write
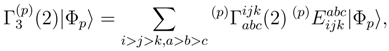
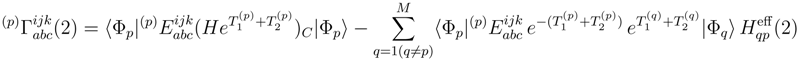
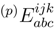 represents the hermitian adjoint to
represents the hermitian adjoint to 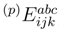 ). In the case of the complete model space 0, considered here, at least one index among i, j, k, a, b, c in Eqs. (53) and (54) must be inactive. and estimates entering the MRMBPT(2) formula by their SUMRCCSD values, as a source of wave function |Ψµ〉 in the MM-SUCC(2,3) energy expressions, Eqs. (48) and (49). The MRMBPT(2) wave function is the lowest-order MRMBPT wave function that contains information about the
). In the case of the complete model space 0, considered here, at least one index among i, j, k, a, b, c in Eqs. (53) and (54) must be inactive. and estimates entering the MRMBPT(2) formula by their SUMRCCSD values, as a source of wave function |Ψµ〉 in the MM-SUCC(2,3) energy expressions, Eqs. (48) and (49). The MRMBPT(2) wave function is the lowest-order MRMBPT wave function that contains information about the 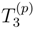 cluster components. The use of wave functions |Ψµ〉 of this type in Eqs. (48) and (49) would lead to a multi-reference extension of the recently proposed completely renormalized CCSD(T) [CR-CCSD(T)] method [7,26,117,118,119,121,132,133]. The spectacular successes of the single-reference CR-CCSD(T) approach in calculations of groundstate PESs involving bond breaking, where the standard CCSD(T) approach completely fails, suggest that the multi-reference analog of the CR-CCSD(T) approach, obtained by inserting the MRMBPT(2)-like wave functions |Ψµ〉 in Eqs. (48) and (49), may provide excellent results, particularly in difficult situations in which the use of the low-order MRMBPT theory alone to estimate the higher-order (e.g., ) effects is not entirely appropriate due to the presence of intruder states.
cluster components. The use of wave functions |Ψµ〉 of this type in Eqs. (48) and (49) would lead to a multi-reference extension of the recently proposed completely renormalized CCSD(T) [CR-CCSD(T)] method [7,26,117,118,119,121,132,133]. The spectacular successes of the single-reference CR-CCSD(T) approach in calculations of groundstate PESs involving bond breaking, where the standard CCSD(T) approach completely fails, suggest that the multi-reference analog of the CR-CCSD(T) approach, obtained by inserting the MRMBPT(2)-like wave functions |Ψµ〉 in Eqs. (48) and (49), may provide excellent results, particularly in difficult situations in which the use of the low-order MRMBPT theory alone to estimate the higher-order (e.g., ) effects is not entirely appropriate due to the presence of intruder states.4 The State-Universal Multi-Reference Coupled-Cluster Method with Perturbative Description of Core-Virtual Excitations: The SUMRCCSD(1) Approach
do not depend on the reference label p [38]. It is, therefore, quite reasonable to introduce an approximation, in which all core–virtual amplitudes of the SUMRCC theory are approximated by their first-order MRMBPT estimates. This new approximation, referred to as the SUMRCCSD(1) method [59], is discussed in this section.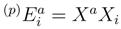 and
and 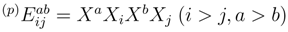 be the excitation operators generating the singly and doubly excited configurations relative to reference |Φp〉. In terms of these operators, the singly and doubly excited clusters of the SUMRCCSD approach take the usual form,
be the excitation operators generating the singly and doubly excited configurations relative to reference |Φp〉. In terms of these operators, the singly and doubly excited clusters of the SUMRCCSD approach take the usual form,
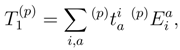
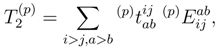
 and
and  are the singly and doubly excited cluster amplitudes obtained by solving the SUMRCCSD equations. Since we are assuming here that 0 is complete, the operators
are the singly and doubly excited cluster amplitudes obtained by solving the SUMRCCSD equations. Since we are assuming here that 0 is complete, the operators 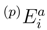 and
and  and the corresponding cluster amplitudes and must carry at least one inactive (i.e., core or virtual) spin-orbital label. and values depend on label p. Indeed, suppose the spin-orbitals i, j, . . . and a, b, . . . (commonly designated as the ρ, σ, . . . spin-orbitals) are obtained in the canonical Hartree-Fock calculations and suppose the reference |Φ1〉 is the ground-state Hartree-Fock determinant. Let us construct the remaining reference configurations |Φp〉, p = 2, . . . , M, in a usual way by choosing some occupied and some unoccupied spin-orbitals in |Φ1〉 as active spin-orbitals and by promoting active electrons (electrons occupying active spin-orbitals in |Φ1〉) to active spin-orbitals that are unoccupied in |Φ1〉. Let us also introduce the Fock matrix elements
and the corresponding cluster amplitudes and must carry at least one inactive (i.e., core or virtual) spin-orbital label. and values depend on label p. Indeed, suppose the spin-orbitals i, j, . . . and a, b, . . . (commonly designated as the ρ, σ, . . . spin-orbitals) are obtained in the canonical Hartree-Fock calculations and suppose the reference |Φ1〉 is the ground-state Hartree-Fock determinant. Let us construct the remaining reference configurations |Φp〉, p = 2, . . . , M, in a usual way by choosing some occupied and some unoccupied spin-orbitals in |Φ1〉 as active spin-orbitals and by promoting active electrons (electrons occupying active spin-orbitals in |Φ1〉) to active spin-orbitals that are unoccupied in |Φ1〉. Let us also introduce the Fock matrix elements
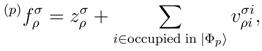
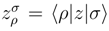 and
and 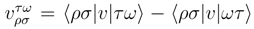 are the one-electron and antisymmetrized two-electron molecular integrals defining the electronic Hamiltonian,
are the one-electron and antisymmetrized two-electron molecular integrals defining the electronic Hamiltonian,
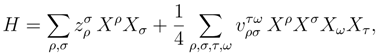
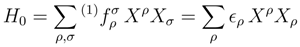

 and , respectively, are
and , respectively, are
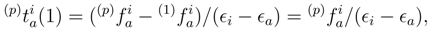

 = 0 (in general, for the canonical Hartree-Fock spinorbitals, the p = 1 matrix elements
= 0 (in general, for the canonical Hartree-Fock spinorbitals, the p = 1 matrix elements 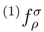 vanish unless ρ = σ). Clearly, the values of (1) and (1) depend on p, since different spin-orbitals are occupied in different reference configurations |Φp〉. In addition, the (1) amplitudes vanish for p = 1 and are, in general, nonzero for p > 1. The order-by-order analysis of the SUMRCCSD method demonstrates that differences between cluster amplitudes associated with different references |Φp〉 are even larger in higher orders of MRMBPT [38]. and amplitudes are, in general, p-dependent and should be treated as independent parameters in the process of solving the SUMRCCSD equations, the first-order core–virtual biexcited amplitudes,
vanish unless ρ = σ). Clearly, the values of (1) and (1) depend on p, since different spin-orbitals are occupied in different reference configurations |Φp〉. In addition, the (1) amplitudes vanish for p = 1 and are, in general, nonzero for p > 1. The order-by-order analysis of the SUMRCCSD method demonstrates that differences between cluster amplitudes associated with different references |Φp〉 are even larger in higher orders of MRMBPT [38]. and amplitudes are, in general, p-dependent and should be treated as independent parameters in the process of solving the SUMRCCSD equations, the first-order core–virtual biexcited amplitudes, 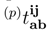 (1), do not depend on label p. We have,
(1), do not depend on label p. We have,
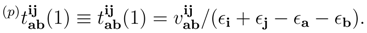 , we simply estimate their values using the first-order MRMBPT expression, Eq. (64) [59]. The first-order core–virtual monoexcited amplitudes
, we simply estimate their values using the first-order MRMBPT expression, Eq. (64) [59]. The first-order core–virtual monoexcited amplitudes 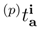 (1) depend on p, but they are usually much smaller than the (1) amplitudes, so that we may simply neglect the core–virtual (1) amplitudes altogether and invoke an additional approximation in which [59]
(1) depend on p, but they are usually much smaller than the (1) amplitudes, so that we may simply neglect the core–virtual (1) amplitudes altogether and invoke an additional approximation in which [59]
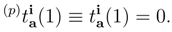
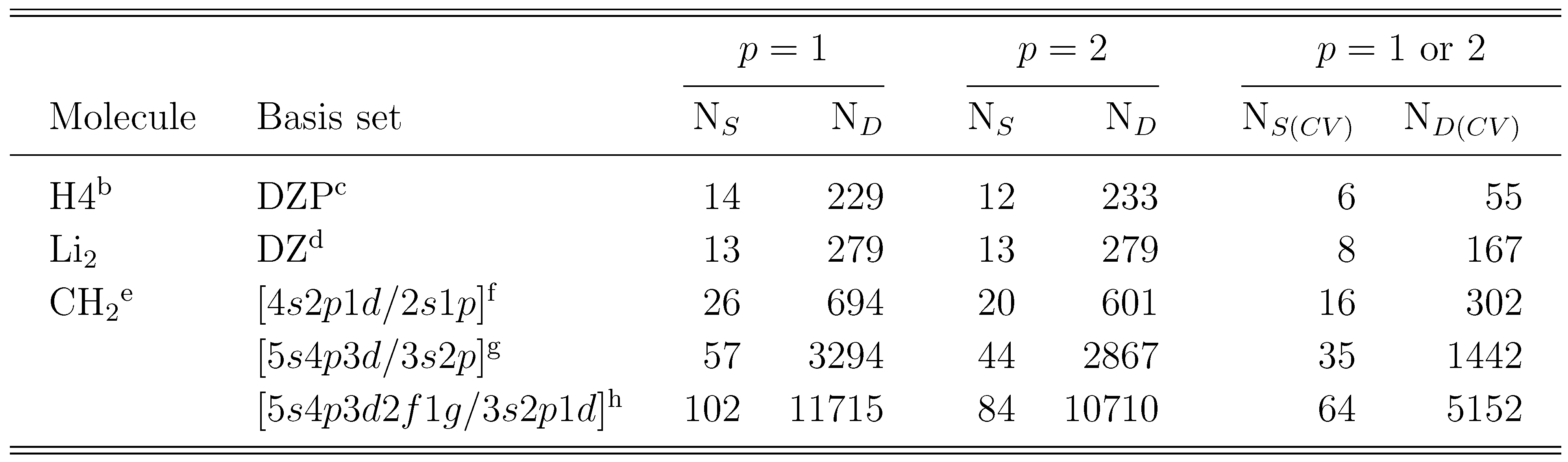
 are simply ignored, so that the number of equations and the number of unknown cluster amplitudes that need to be determined in the iterative procedure are identical. As shown in Table 1, this considerably reduces the computer effort involved, since in the SUMRCCSD(1) method we only consider a relatively small subset of all SUMRCCSD amplitude equations corresponding to single and double excitations that carry at least one active index, i.e.,
are simply ignored, so that the number of equations and the number of unknown cluster amplitudes that need to be determined in the iterative procedure are identical. As shown in Table 1, this considerably reduces the computer effort involved, since in the SUMRCCSD(1) method we only consider a relatively small subset of all SUMRCCSD amplitude equations corresponding to single and double excitations that carry at least one active index, i.e., 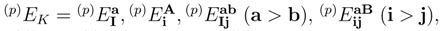
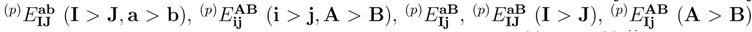 . We use these equations to determine the relatively small subset of all and amplitudes that carry at least one active index. Even for small systems, such as the methylene molecule, and even for the simple two-reference (M = 2) singlet case, the savings in the computer effort are substantial. For example, the total number of spin- and symmetry-adapted cluster amplitudes used in the two-reference OSA SUMRCCSD calculations for the the lowest two 1A1 states of methylene, as described by the double-zeta plus polarization (DZP) basis set of Refs. 142, 143, is 1341 (720 for |Φ1〉 and 621 for |Φ2); see Table 1). The number of core–virtual amplitudes for each reference is in this case 318, so that the total number of core–virtual amplitudes considered in the two-reference OSA SUMRCCSD calculations for the DZP methylene molecule is 636. These 636 amplitudes are estimated in the SUMRCCSD(1) method by the first-order MRMBPT expression, Eq. (64), and by Eq. (65), and only the remaining 705 cluster amplitudes that carry at least one active orbital index are determined iteratively by solving the relevant subset of all SUMRCCSD equations. A similar ∼ 50 % reduction in the number of amplitudes that have to be determined in the SUMRCCSD(1) iterative procedure characterizes the calculations for methylene employing a larger [5s4p3d2f1g/3s2p1d] basis set of Ref. 144 (cf., also, Ref. 52). In this case, the total number of core–virtual amplitudes considered in the two-reference OSA SUMRCCSD calculations is 10432, while the number of all singly and doubly excited cluster amplitudes is 22611 (see Table 1). The 10432 core–virtual amplitudes are estimated in the SUMRCCSD(1) calculations via Eqs. (64) and (65), whereas the remaining 12179 amplitudes are determined iteratively. This significant reduction in the number of cluster amplitudes that have to be determined in the SUMRCCSD(1) iterative procedure would be even larger for larger many-electron systems, since the number of core–virtual amplitudes increases with the number of core electrons.
. We use these equations to determine the relatively small subset of all and amplitudes that carry at least one active index. Even for small systems, such as the methylene molecule, and even for the simple two-reference (M = 2) singlet case, the savings in the computer effort are substantial. For example, the total number of spin- and symmetry-adapted cluster amplitudes used in the two-reference OSA SUMRCCSD calculations for the the lowest two 1A1 states of methylene, as described by the double-zeta plus polarization (DZP) basis set of Refs. 142, 143, is 1341 (720 for |Φ1〉 and 621 for |Φ2); see Table 1). The number of core–virtual amplitudes for each reference is in this case 318, so that the total number of core–virtual amplitudes considered in the two-reference OSA SUMRCCSD calculations for the DZP methylene molecule is 636. These 636 amplitudes are estimated in the SUMRCCSD(1) method by the first-order MRMBPT expression, Eq. (64), and by Eq. (65), and only the remaining 705 cluster amplitudes that carry at least one active orbital index are determined iteratively by solving the relevant subset of all SUMRCCSD equations. A similar ∼ 50 % reduction in the number of amplitudes that have to be determined in the SUMRCCSD(1) iterative procedure characterizes the calculations for methylene employing a larger [5s4p3d2f1g/3s2p1d] basis set of Ref. 144 (cf., also, Ref. 52). In this case, the total number of core–virtual amplitudes considered in the two-reference OSA SUMRCCSD calculations is 10432, while the number of all singly and doubly excited cluster amplitudes is 22611 (see Table 1). The 10432 core–virtual amplitudes are estimated in the SUMRCCSD(1) calculations via Eqs. (64) and (65), whereas the remaining 12179 amplitudes are determined iteratively. This significant reduction in the number of cluster amplitudes that have to be determined in the SUMRCCSD(1) iterative procedure would be even larger for larger many-electron systems, since the number of core–virtual amplitudes increases with the number of core electrons.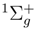 states of the Li2 molecule, as described by the double zeta (DZ) basis set of Refs. 148, 149 (see Table 3), and the lowest two 1A1 states of methylene, as described by the DZP basis set [142,143] and the [5s4p3d/3s2p] [52] and [5s4p3d2f1g/3s2p1d] atomic natural orbital [144] basis sets (see Table 4). The results of the SUMRCCSD(1) calculations for H4 and CH2 were reported earlier [59]. The results for Li2 are new.
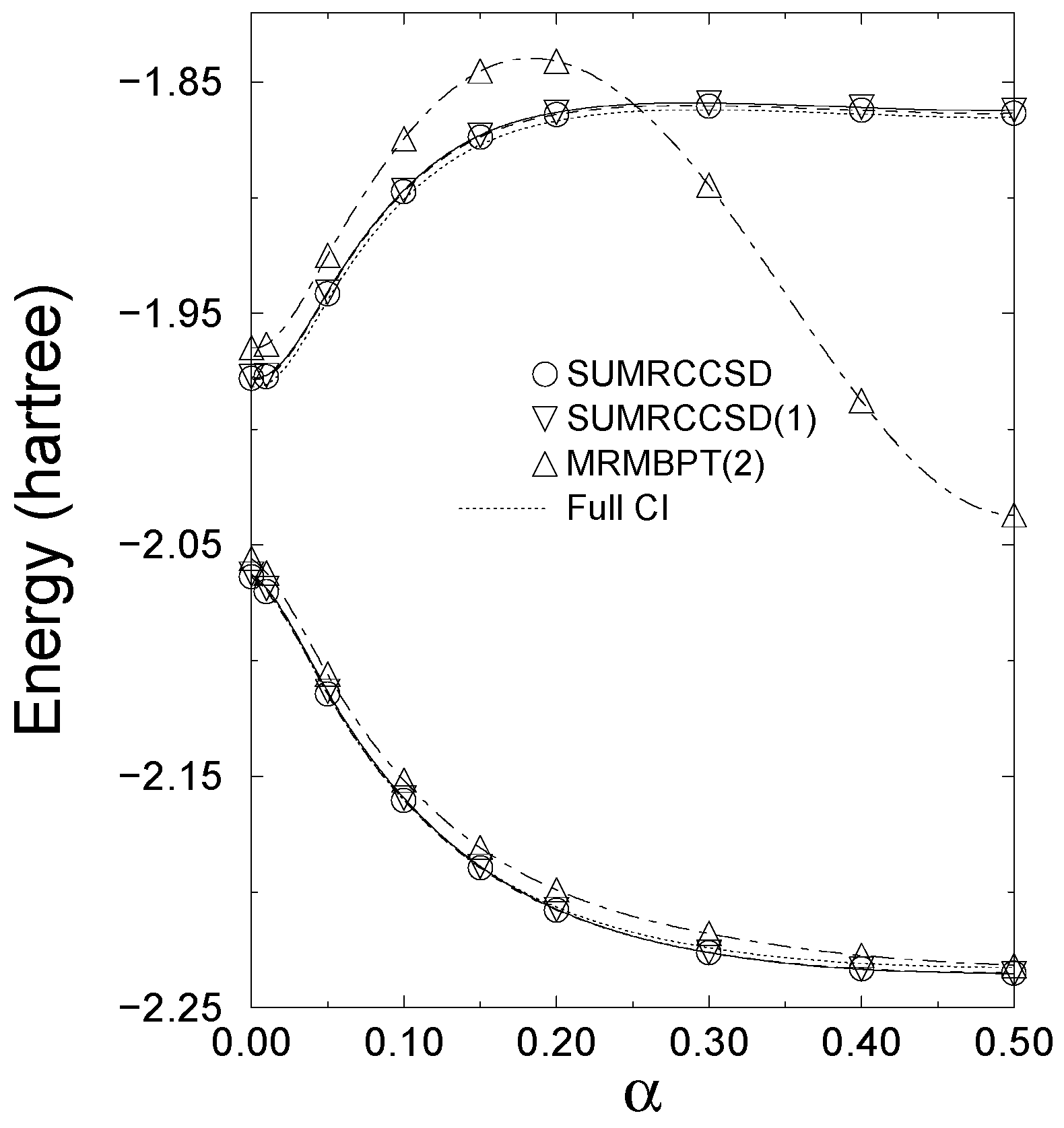
states of the Li2 molecule, as described by the double zeta (DZ) basis set of Refs. 148, 149 (see Table 3), and the lowest two 1A1 states of methylene, as described by the DZP basis set [142,143] and the [5s4p3d/3s2p] [52] and [5s4p3d2f1g/3s2p1d] atomic natural orbital [144] basis sets (see Table 4). The results of the SUMRCCSD(1) calculations for H4 and CH2 were reported earlier [59]. The results for Li2 are new.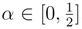 . The α = 0 and
. The α = 0 and  limits correspond to square and linear conformations, respectively [146].
limits correspond to square and linear conformations, respectively [146].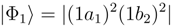
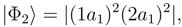 symmetry are dominated by the RHF configuration
symmetry are dominated by the RHF configuration
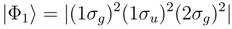
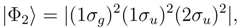 states of Li2 are only slightly above the corresponding SUMRCCSD energies [the difference between the SUMRCCSD(1) and SUMRCCSD energies is ∼ 2.5 millihartree, independently of the value of R]. In consequence, the errors in the SUMRCCSD(1) results, relative to full CI, are very small (2.7–2.8 millihartree for the ground state and 3.1–3.2 millihartree for the first-excited state). The errors in the 1 → 2 vertical excitation energies (designated by ∆) obtained with the SUMRCCSD(1) method, relative to full CI, are 0.4–0.5 millihartree, independently of the value of R. The SUMRCCSD results for the 1 → 2 excitation energies are virtually identical to those obtained with the simpler SUMRCCSD(1) approach. The two-reference MRMBPT(2) method works reasonably well in the quasi-degenerate region corresponding to larger R values, where the lowest two states are dominated by configurations (68) and (69), but the SUMRCCSD(1) results are much more accurate [cf., e.g., the 12.8 millihartree error in the MRMBPT(2) result for the vertical excitation energy ∆ at R = 2Re with the 0.3–0.4 millihartree errors obtained at the same value of R with the SUMRCCSD and SUMRCCSD(1) approaches]. In the R ≈ Re region, the ground state is dominated by the RHF configuration |Φ1〉, Eq. (68), but the first-excited state of the symmetry has significant contributions from configurations belonging to . As a result, the error in the MRMBPT(2) value for the vertical excitation energy ∆ is as large as 54.0 millihartree at R = Re. Remarkably enough, the errors in the SUMRCCSD and SUMRCCSD(1) results for the 1 → 2 excitation energy are as little as 0.5 millihartree at R = Re. This clearly shows that we can tremendously benefit from incorporating the MRMBPT ideas into the SUMRCCSD scheme.
states of Li2 are only slightly above the corresponding SUMRCCSD energies [the difference between the SUMRCCSD(1) and SUMRCCSD energies is ∼ 2.5 millihartree, independently of the value of R]. In consequence, the errors in the SUMRCCSD(1) results, relative to full CI, are very small (2.7–2.8 millihartree for the ground state and 3.1–3.2 millihartree for the first-excited state). The errors in the 1 → 2 vertical excitation energies (designated by ∆) obtained with the SUMRCCSD(1) method, relative to full CI, are 0.4–0.5 millihartree, independently of the value of R. The SUMRCCSD results for the 1 → 2 excitation energies are virtually identical to those obtained with the simpler SUMRCCSD(1) approach. The two-reference MRMBPT(2) method works reasonably well in the quasi-degenerate region corresponding to larger R values, where the lowest two states are dominated by configurations (68) and (69), but the SUMRCCSD(1) results are much more accurate [cf., e.g., the 12.8 millihartree error in the MRMBPT(2) result for the vertical excitation energy ∆ at R = 2Re with the 0.3–0.4 millihartree errors obtained at the same value of R with the SUMRCCSD and SUMRCCSD(1) approaches]. In the R ≈ Re region, the ground state is dominated by the RHF configuration |Φ1〉, Eq. (68), but the first-excited state of the symmetry has significant contributions from configurations belonging to . As a result, the error in the MRMBPT(2) value for the vertical excitation energy ∆ is as large as 54.0 millihartree at R = Re. Remarkably enough, the errors in the SUMRCCSD and SUMRCCSD(1) results for the 1 → 2 excitation energy are as little as 0.5 millihartree at R = Re. This clearly shows that we can tremendously benefit from incorporating the MRMBPT ideas into the SUMRCCSD scheme.
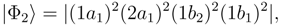
5 Summary
Acknowledgements
References
- Čížek, J. J. Chem. Phys. 1966, 45, 4256.
- Čížek, J. Adv. Chem. Phys. 1969, 14, 35.
- Čížek, J.; Paldus, J. Int. J. Quantum Chem. 1971, 5, 359.
- Paldus, J. Methods in Computational Molecular Physics; Wilson, S., Diercksen, G.H.F., Eds.; NATO Advanced Study Institute, Series B: Physics; Vol. 293, Plenum: New York, 1992; pp. 99–194. [Google Scholar]
- Bartlett, R.J. Modern Electronic Structure Theory; Yarkony, D.R., Ed.; World Scientific: Singapore, 1995; Part I; pp. 1047–1131. [Google Scholar]
- Paldus, J.; Li, X. Adv. Chem. Phys. 1999, 110, 1.
- Piecuch, P.; Kowalski, K. Computational Chemistry: Reviews of Current Trends; Leszczyński, J., Ed.; World Scientific: Singapore, 2000. [Google Scholar]
- Bloch, C. Nucl. Phys. 1958, 6, 329.
- Jørgensen, F. Mol. Phys. 1975, 29, 1137.
- Soliverez, C.E. Phys. Rev. A 1981, 24, 4.
- Durand, Ph. Phys. Rev. A 1983, 28, 3184.
- Hubartubise, V.; Freed, K.F. Adv. Chem. Phys. 1993, 80, 465.
- Oliphant, N.; Adamowicz, L. J. Chem. Phys. 1991, 94, 1229.
- Oliphant, N.; Adamowicz, L. J. Chem. Phys. 1992, 96, 3739.
- Oliphant, N.; Adamowicz, L. Int. Rev. Phys. Chem. 1993, 12, 339.
- Piecuch, P.; Oliphant, N.; Adamowicz, L. J. Chem. Phys. 1993, 99, 1875.
- Piecuch, P.; Adamowicz, L. J. Chem. Phys. 1994, 100, 5792.
- Piecuch, P.; Adamowicz, L. Chem. Phys. Lett. 1994, 221, 121.
- Piecuch, P.; Adamowicz, L. J. Chem. Phys. 1995, 102, 898.
- Ghose, K.B.; Adamowicz, L. J. Chem. Phys. 1995, 103.
- Ghose, K.B.; Piecuch, P.; Adamowicz, L. J. Chem. Phys. 1995, 103, 9331.
- Ghose, K.B.; Piecuch, P.; Pal, S.; Adamowicz, L. J. Chem. Phys. 1996, 104, 6582.
- Adamowicz, L.; Piecuch, P.; Ghose, K.B. Mol. Phys. 1998, 94, 225.
- Piecuch, P.; Kucharski, S.A.; Bartlett, R.J. J. Chem. Phys. 1999, 110, 6103.
- Piecuch, P.; Kucharski, S.A.; Špirko, V. J. Chem. Phys. 1999, 111, 6679.
- Kowalski, K.; Piecuch, P. Chem. Phys. Lett. 2001, 344, 165.
- Kowalski, K.; Piecuch, P. J. Chem. Phys. 2000, 113, 8490.
- Kowalski, K.; Piecuch, P. J. Chem. Phys. 2001, 115, 643.
- Kowalski, K.; Piecuch, P. Chem. Phys. Lett. 2001, 347, 237.
- Mahapatra, U.S.; Datta, B.; Mukherjee, D. Mol. Phys. 1998, 94, 157.
- Mahapatra, U.S.; Datta, B.; Mukherjee, D. J. Chem. Phys. 1999, 110, 6171.
- Mach, P.; Mášsik, J.; Urban, J.; Hubačc, I. Mol. Phys. 1998, 94, 173.
- Mášsik, J.; Hubač, I. Adv. Quantum Chem. 1999, 31, 75.
- Pittner, J.; Nachtigall, P.; Čársky, P.; Mášsik, J.; Hubač, I. J. Chem. Phys. 1999, 110, 10275.
- Hubač, I.; Pittner, J.; Čársky, P. J. Chem. Phys. 2000, 112, 8779.
- Sancho-Garcia, J.C.; Pittner, J.; Čársky, P.; Hubač, I. J. Chem. Phys. 2000, 112, 8785.
- Pittner, J.; Nachtigall, P.; Čársky, P.; Hubač, I. J. Phys. Chem. A 2001, 105, 1354.
- Jeziorski, B.; Monkhorst, H.J. Phys. Rev. A 1981, 24, 1668.
- Jeziorski, B.; Paldus, J. J. Chem. Phys. 1988, 88, 5673.
- Meissner, L.; Jankowski, K.; Wasilewski, J. Int. J. Quantum Chem. 1988, 34, 535.
- Paldus, J.; Pylypow, L.; Jeziorski, B. Many-Body Methods in Quantum Chemistry; Kaldor, U., Ed.; Lecture Notes in Chemistry; Vol. 52, Springer: Berlin, 1989; pp. 151–170. [Google Scholar]
- Kucharski, S.A.; Bartlett, R.J. J. Chem. Phys. 1991, 95, 8227.
- Balková, A.; Kucharski, S.A.; Meissner, L.; Bartlett, R.J. Theor. Chim. Acta 1991, 80, 335.
- Balková, A.; Kucharski, S.A.; Bartlett, R.J. Chem. Phys. Lett. 1991, 182, 511.
- Piecuch, P.; Paldus, J. Theor. Chim. Acta 1992, 83, 69.
- Paldus, J.; Piecuch, P.; Jeziorski, B.; Pylypow, L. Recent Progress in Many-Body Theories; Ainsworthy, T.L., Campbell, C.E., Clements, B.E., Krotschek, E., Eds.; Plenum: New York, 1992. [Google Scholar]
- Paldus, J.; Piecuch, P.; Pylypow, L.; Jeziorski, B. Phys. Rev. A 1993, 47, 2738. [PubMed]
- Piecuch, P.; Toboła, R.; Paldus, J. Chem. Phys. Lett. 1993, 210, 243.
- Piecuch, P.; Paldus, J. Phys. Rev. A 1994, 49, 3479. [PubMed]
- Piecuch, P.; Paldus, J. J. Chem. Phys. 1994, 101, 5875.
- Li, X.; Piecuch, P.; Paldus, J. Chem. Phys. Lett. 1994, 224, 267.
- Piecuch, P.; Li, X.; Paldus, J. Chem. Phys. Lett. 1994, 230, 377.
- Balková, A.; Bartlett, R.J. J. Chem. Phys. 1994, 101, 8972.
- Piecuch, P.; Paldus, J. J. Phys. Chem. 1995, 99, 15354.
- Berkovic, S.; Kaldor, U. Chem. Phys. Lett. 1992, 42, 199.
- Berkovic, S.; Kaldor, U. J. Chem. Phys. 1993, 98, 3090.
- Kowalski, K.; Piecuch, P. Phys. Rev. A 2000, 61, 052506.
- Piecuch, P.; Landman, J.I. Parallel Comp. 2000, 26, 913.
- Kowalski, K.; Piecuch, P. Chem. Phys. Lett. 2001, 334, 89.
- Coester, F. Lectures in Theoretical Physics; Mahanthappa, K.T., Brittin, W.E., Eds.; Gordon and Breach: New York, 1969. [Google Scholar]
- Lindgren, I. J. Phys. B 1974, 7, 2441.
- Mukherjee, D.; Moitra, R.K.; Mukhopadhyay, A. Mol. Phys. 1975, 30, 1861.
- Offerman, R.; Ey, W.; Kümmel, H. Nucl. Phys. A 1976, 273, 349.
- Lindgren, I. Int. J. Quantum Chem. Symp. 1978, 12, 33.
- Mukhopadhyay, A.; Moitra, R.K.; Mukherjee, D. J. Phys. B 1979, 12, 1.
- Kutzelnigg, W. J. Chem. Phys. 1982, 77, 3081.
- Pal, S.; Prasad, M.D.; Mukherjee, D. Theor. Chim. Acta 1983, 62, 523.
- Haque, M.A.; Mukherjee, D. J. Chem. Phys. 1984, 80, 5058.
- Kutzelnigg, W. J. Chem. Phys. 1984, 80, 822.
- Stolarczyk, L.; Monkhorst, H.J. Phys. Rev. A 1985, 32, 725. [PubMed]
- Stolarczyk, L.; Monkhorst, H.J. Phys. Rev. A 1985, 32, 743. [PubMed]
- Haque, A.; Kaldor, U. Chem. Phys. Lett. 1985, 117, 347.
- Lindgren, I. Phys. Scr. 1985, 32, 291.
- Lindgren, I. Phys. Scr. 1985, 32, 611.
- Haque, A.; Kaldor, U. Int. J. Quantum Chem. 1986, 29, 425.
- Mukherjee, D. Chem. Phys. Lett. 1986, 125, 207.
- Kaldor, U. J. Chem. Phys. 1987, 87, 467.
- Lindgren, I.; Mukherjee, D. Phys. Rep. 1987, 151, 93.
- Kutzelnigg, W.; Mukherjee, D.; Koch, S. J. Chem. Phys. 1987, 87, 5902.
- Mukherjee, D.; Kutzelnigg, W.; Koch, S. J. Chem. Phys. 1987, 87, 5911.
- Pal, S.; Rittby, M.; Bartlett, R.J.; Sinha, D.; Mukherjee, D. Chem. Phys. Lett. 1987, 137, 273.
- Ghose, K.B.; Pal, S. J. Chem. Phys. 1988, 97, 3863.
- Pal, S.; Rittby, M.; Bartlett, R.J.; Sinha, D.; Mukherjee, D. J. Chem. Phys. 1988, 88, 4357.
- Mukherjee, D.; Pal, S. Adv. Quantum Chem. 1989, 20, 291.
- Jeziorski, B.; Paldus, J. J. Chem. Phys. 1989, 90, 2714.
- Lindgren, I. J. Phys. B 1991, 24, 1143.
- Kaldor, U. Theor. Chim. Acta 1991, 80, 427.
- Stanton, J.F.; Bartlett, R.J.; Rittby, C.M.L. J. Chem. Phys. 1992, 97, 5560.
- Malinowski, P.; Jankowski, K. J. Phys. B 1993, 26, 3035.
- Jankowski, K.; Malinowski, P. J. Phys. B 1994, 27, 829.
- Malinowski, P.; Jankowski, K. Phys. Rev. A 1995, 51, 4583. [PubMed]
- Hughes, S.R.; Kaldor, U. Int. J. Quantum Chem. 1995, 55, 127.
- Meissner, L. J. Chem. Phys. 1995, 103, 8014.
- Roeselova, M.; Jacoby, G.; Kaldor, U.; Jungwirth, P. Chem. Phys. Lett. 1998, 293, 309.
- Eliav, E.; Kaldor, U.; Hess, B.A. J. Chem. Phys. 1998, 108, 3409.
- Landau, A.; Eliav, E.; Kaldor, U. Chem. Phys. Lett. 1999, 313, 399.
- Bernholdt, D.E.; Bartlett, R.J. Adv. Quantum Chem. 1999, 34, 271.
- Jankowski, K.; Paldus, J.; Grabowski, I.; Kowalski, K. J. Chem. Phys. 1994, 101, 1759–Erratum.
- Jankowski, K.; Paldus, J.; Grabowski, I.; Kowalski, K. J. Chem. Phys. 1994, 101, 3085.
- Nooijen, M.; Bartlett, R.J. J. Chem. Phys. 1997, 106, 6441.
- Nooijen, M.; Bartlett, R.J. J. Chem. Phys. 1997, 106, 6812.
- Paldus, J.; Adams, B.; Čížek, J. Int. J. Quantum Chem. 1977, 11, 813.
- Paldus, J. J. Chem. Phys. 1977, 67, 303.
- Adams, B.G.; Paldus, J. Phys. Rev. A 1979, 20, 1.
- Piecuch, P.; Paldus, J. Int. J. Quantum Chem. 1989, 36, 429.
- Piecuch, P.; Paldus, J. Theor. Chim. Acta 1990, 78, 65.
- Jankowski, K.; Meissner, L.; Rubiniec, K. J. Molec. Struct.: THEOCHEM 2001, 547, 55.
- Li, X.; Paldus, J. J. Chem. Phys. 1994, 101, 8812.
- Jensen, P.; Bunker, P.R. J. Chem. Phys. 1988, 89, 1327.
- Shamasundar, K.R.; Pal, S. J. Chem. Phys. 2001, 115, 1979–Erratum.
- Kowalski, K.; Piecuch, P. Int. J. Quantum Chem. 2000, 80, 757.
- Brandow, B.H. Rev. Mod. Phys. 1967, 39, 771.
- Lindgren, I. J. Phys. B 1974, 7, 2441.
- Lindgren, I.; Morrison, J. Atomic Many-Body Theory; Springer: Berlin, 1982. [Google Scholar]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. Chem. Phys. Lett. 1989, 157, 479.
- Kucharski, S.A.; Bartlett, R.J. J. Chem. Phys. 1998, 108, 9221.
- Kowalski, K.; Piecuch, P. J. Mol. Struct.: THEOCHEM 2001, 547, 191.
- Kowalski, K.; Piecuch, P. J. Chem. Phys. 2000, 113, 18.
- Kowalski, K.; Piecuch, P. J. Chem. Phys. 2000, 113, 5644.
- Piecuch, P.; Kowalski, K.; Pimienta, I.S.O. Int. J. Mol. Sci. 2002, 3, 475.
- Piecuch, P.; Kowalski, K.; Pimienta, I.S.O.; Kucharski, S.A. Low-Lying Potential Energy Surfaces; Hoffmann, M.R., Dyall, K.G., Eds.; ACS: Washington, D.C, 2002. [Google Scholar]
- Kowalski, K.; Piecuch, P. J. Chem. Phys. 2001, 115, 2966.
- Kowalski, K.; Piecuch, P. J. Chem. Phys. 2002, 116, 7411.
- Hirao, K. Int. J. Quantum Chem. Symp. 1992, 26, 517.
- Meissner, L.; Nooijen, M. J. Chem. Phys. 1995, 102, 9604.
- Meissner, L.; Kucharski, S.A.; Bartlett, R.J. J. Chem. Phys. 1989, 91, 6187.
- Meissner, L.; Bartlett, R.J. J. Chem. Phys. 1990, 92, 561.
- Geertsen, J.; Rittby, M.; Bartlett, R.J. Chem. Phys. Lett. 1989, 164, 57.
- Stanton, J.F.; Bartlett, R.J. J. Chem. Phys. 1993, 98, 7029.
- Piecuch, P.; Bartlett, R.J. Adv. Quantum Chem. 1999, 34, 295.
- Piecuch, P.; Kucharski, S.A.; Kowalski, K. Chem. Phys. Lett. 2001, 344, 176.
- Piecuch, P.; Kucharski, S.A.; Špírko, V.; Kowalski, K. J. Chem. Phys. 2001, 115, 5796.
- McGuire, M.J.; Kowalski, K.; Piecuch, P. J. Chem. Phys. 2002, 117, XXXX, (in press; the August 22, 2002 issue).
- Meissner, L.; Bartlett, R.J. J. Chem. Phys. 2001, 115, 50.
- Piecuch, P.; Kowalski, K. (unpublished).
- Li, X.; Paldus, J. J. Chem. Phys. 1997, 107, 6257.
- Li, X.; Paldus, J. J. Chem. Phys. 1998, 108, 637.
- Li, X.; Paldus, J. Chem. Phys. Lett. 1998, 286, 145.
- Li, X.; Paldus, J. J. Chem. Phys. 1999, 110, 2844.
- Li, X.; Paldus, J. Mol. Phys. 2000, 98, 1185.
- Li, X.; Paldus, J. J. Chem. Phys. 2000, 113, 9966.
- Bauschlicher, C.W., Jr.; Taylor, P.R. J. Chem. Phys. 1986, 85, 6510.
- Bauschlicher, C.W.; Taylor, P.R. J. Chem. Phys. 1987, 86, 2844.
- Comeau, D.C.; Shavitt, I.; Jensen, P.; Bunker, P.R. J. Chem. Phys. 1989, 90, 6491.
- Landman, J.I.; Piecuch, P. Fortran Forum. 2000, 19, 16. [Google Scholar]
- Jankowski, J.; Paldus, J. Int. J. Quantum Chem. 1980, 17, 1243.
- Paldus, J.; Wormer, P.E.S.; Benard, M. Coll. Czech. Chem. Commun. 1988, 53, 1919.
- Dunning, T.H., Jr.; Hay, P.J. Methods of Electronic Structure Theory; Schaefer, H.F., III, Ed.; Plenum Press: New York, 1977. [Google Scholar]
- Basis set was obtained from the Extensible Computational Chemistry Environment Basis Set Database, Version 11/29/01, as developed and distributed by the Molecular Science Computing Facility, Environmental and Molecular Sciences Laboratory which is part of the Pacific Northwest Laboratory, P.O. Box 999, Richland, Washington 99352, USA, and funded by the U.S. Department of Energy. The Pacific Northwest Laboratory is a multi-program laboratory operated by Battelle Memorial Institute for the U.S. Department of Energy under contract DE-AC06-76RLO 1830. Contact David Feller or Karen Schuchardt for further information.
- Nakano, H. J. Chem. Phys. 1993, 99, 7983.
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; Windus, T.L.; Dupuis, M.; Montgomery, J.A. J. Comput. Chem. 1993, 14, 1347.


 states of the DZ model of Li2 and the SUMRCCSD(1), SUMRCCSD, MRMBPT(2), and full CI values of the 1 → 2 excitation energies (∆, in millihartree) for the equilibrium valueof the Li–Li separation R (R = Re = 5.051 bohr) and two stretches of the Li–Li bond (R = 1.5Re and R = 2Re).
states of the DZ model of Li2 and the SUMRCCSD(1), SUMRCCSD, MRMBPT(2), and full CI values of the 1 → 2 excitation energies (∆, in millihartree) for the equilibrium valueof the Li–Li separation R (R = Re = 5.051 bohr) and two stretches of the Li–Li bond (R = 1.5Re and R = 2Re).


{kind=link}
©2002 by MDPI, Basel, Switzerland. Reproduction for noncommercial purposes permitted.
Share and Cite
Piecuch, P.; Kowalski, K. The State-Universal Multi-Reference Coupled-Cluster Theory: An Overview of Some Recent Advances. Int. J. Mol. Sci. 2002, 3, 676-709. https://doi.org/10.3390/i3060676
Piecuch P, Kowalski K. The State-Universal Multi-Reference Coupled-Cluster Theory: An Overview of Some Recent Advances. International Journal of Molecular Sciences. 2002; 3(6):676-709. https://doi.org/10.3390/i3060676
Chicago/Turabian StylePiecuch, Piotr, and Karol Kowalski. 2002. "The State-Universal Multi-Reference Coupled-Cluster Theory: An Overview of Some Recent Advances" International Journal of Molecular Sciences 3, no. 6: 676-709. https://doi.org/10.3390/i3060676
APA StylePiecuch, P., & Kowalski, K. (2002). The State-Universal Multi-Reference Coupled-Cluster Theory: An Overview of Some Recent Advances. International Journal of Molecular Sciences, 3(6), 676-709. https://doi.org/10.3390/i3060676