
High Concentrations of Circulating 2PY and 4PY—Potential Risk Factor of Cardiovascular Disease in Patients with Chronic Kidney Disease


Abstract
1. Introduction
2. NAD+ Precursors Ingested by Humans
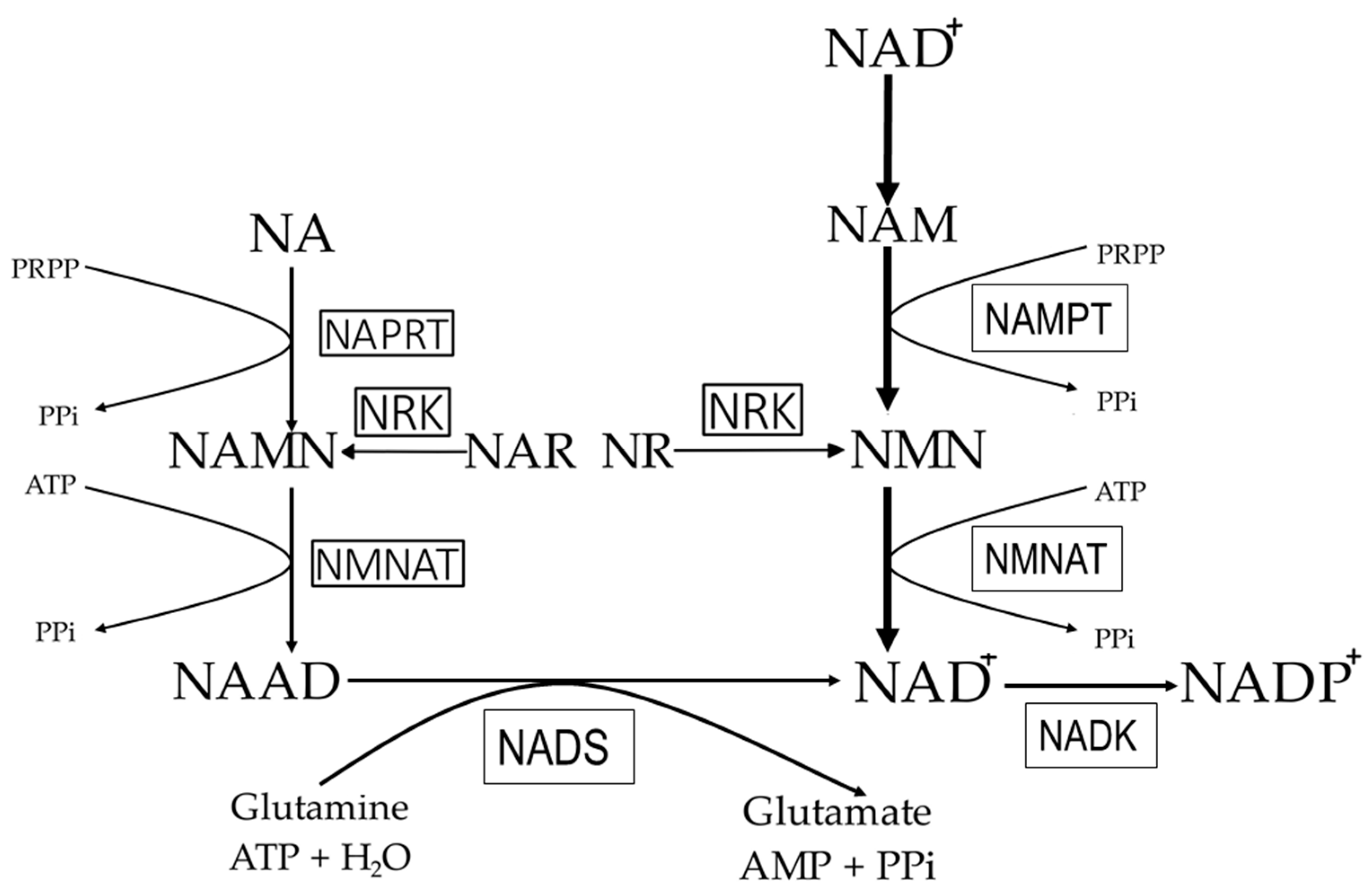
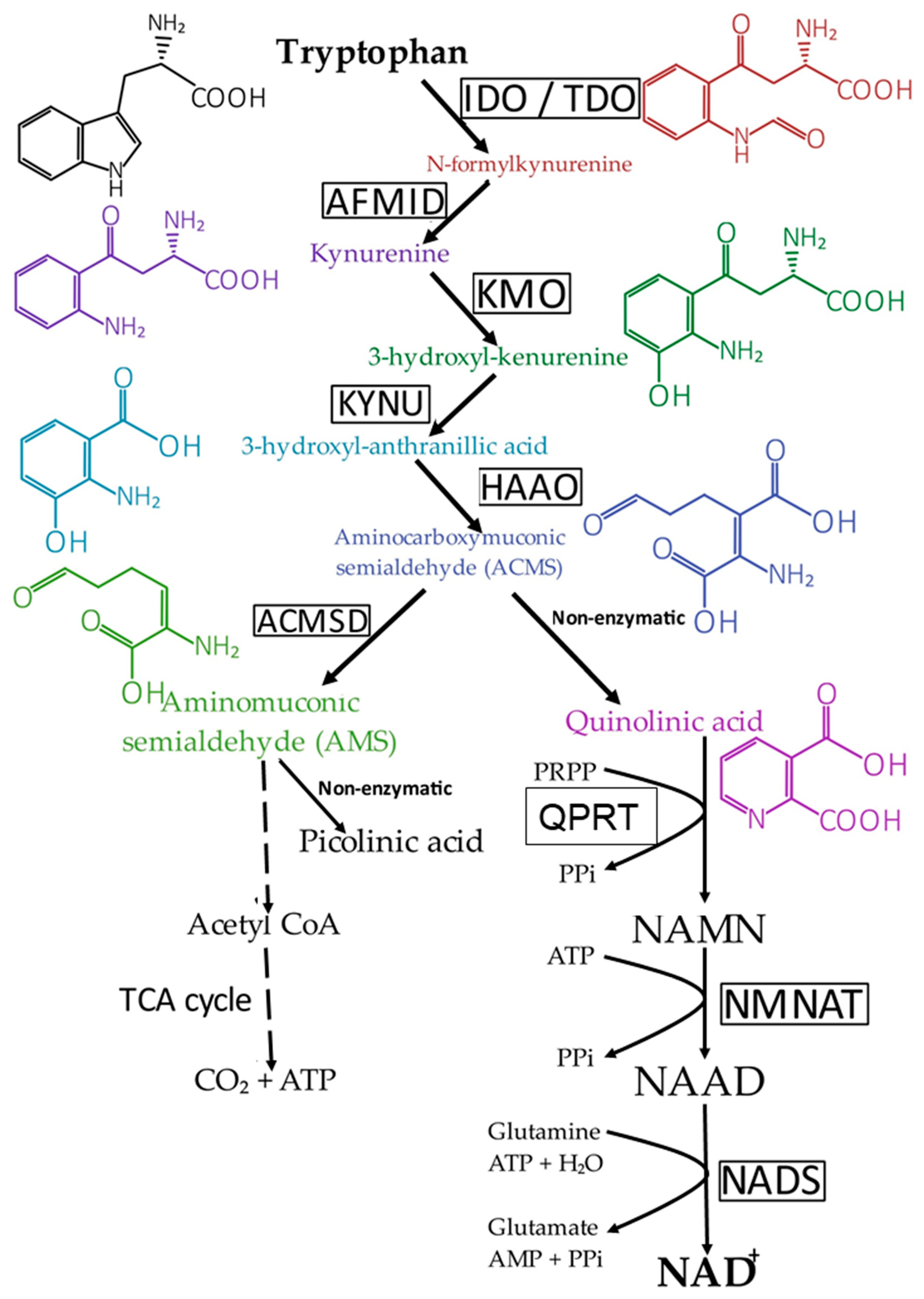
3. NAD+ Synthesis Pathways in Mammals
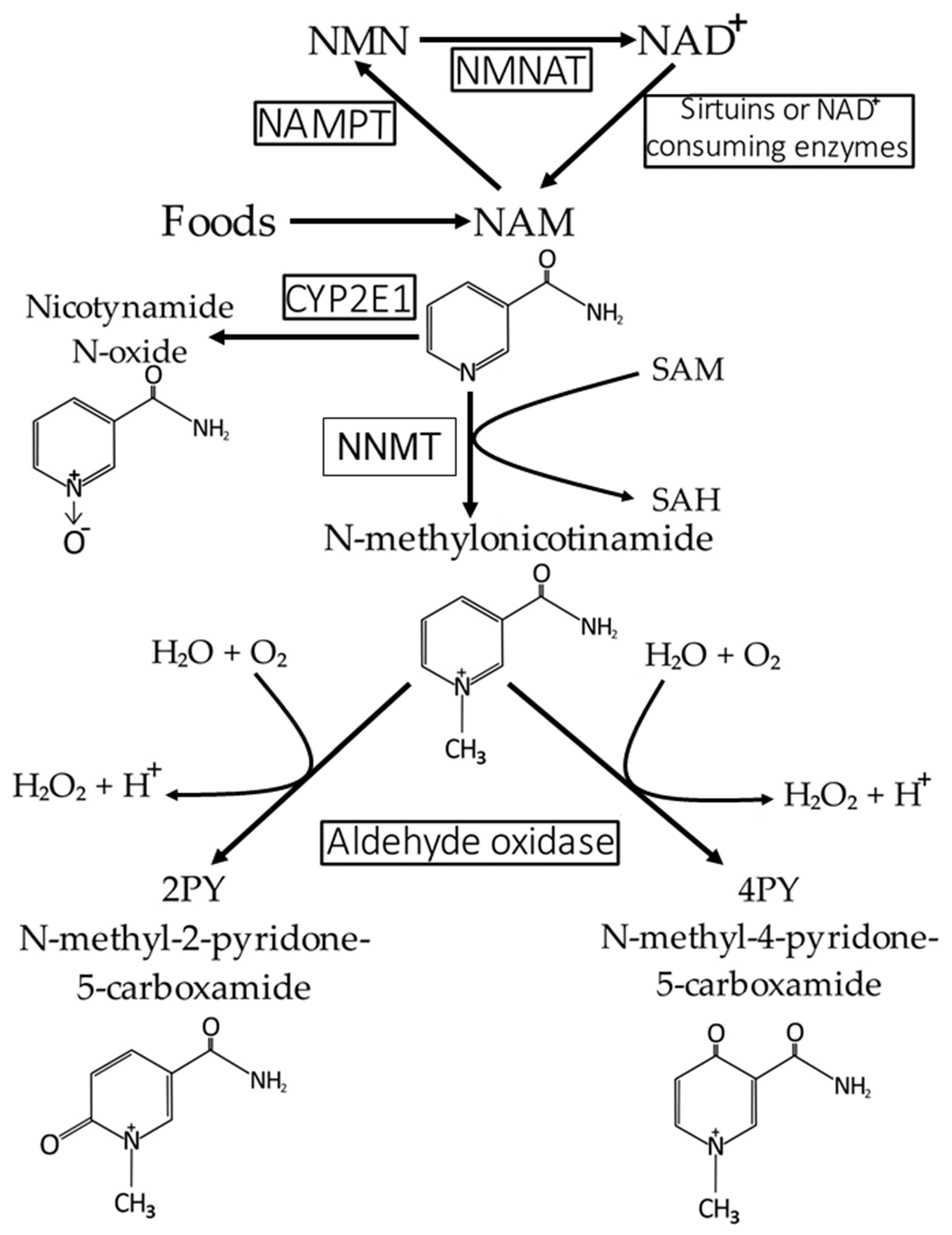
4. NAD+ and NAM Catabolism and Excretion of Formed Catabolites
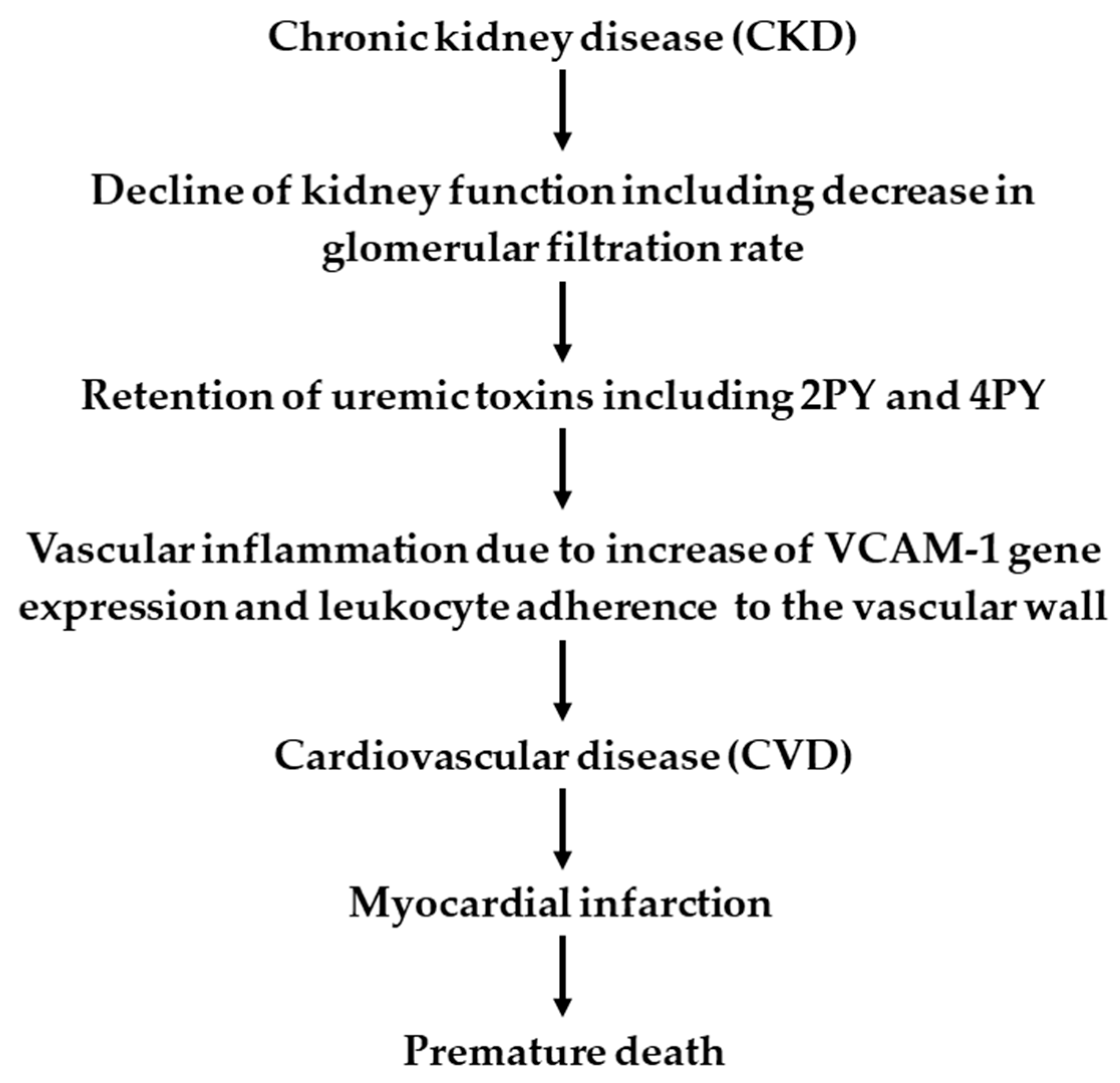
5. Potential Pathophysiological Function of NAD+ Metabolites in CKD Patients
6. Beneficial and Possible Adverse Side Effects of NAD+ Precursors (NAD+ Boosters) Used as Therapeutic Agents or Supplements
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2PY | N-methyl-2-pyrdone-5-carboxamide |
| 4PY | N-methyl-2-4yrdone-5-carboxamide |
| ACMS | aminocarboxymuconic semialhedyde |
| ACMSD | aminocarboxymuconic semialhedyde decarboxylase [EC:4.1.1.45] |
| ADP | adenosine diposphate |
| AMID | N-formylkynureine for-mamidase [EC:3.5.1.9] |
| AMP | adenosine monoposphate |
| AMS | aminomuconic semialdehyde |
| MNAM | N1-methylnicotinamide |
| ATP | adenosine triphosphate |
| CKD | chronic kidney disease |
| COX-2 | cyclooxygenase-2 [EC:1.14.99.1] |
| CYP2E1 | cytochrome 2E1 (P450 cytochrome) [EC:1.14.13.n7] |
| CVD | cardiovascular disease |
| cADPR | cyclic ADP-ribose |
| GPX4 | glutathione peroxidase 4 [EC:1.11.1.12] |
| GSH | glutathione reduced |
| HAAO | 3-hydroxyanthranilate 3,4-di-oxygenase 2 [EC:1.13.11.6] |
| HDAC | histone deacetylase [EC: 3.5.1.98] |
| HDL | High-density lipoprotein |
| IDO | indoleamine2,3-dioxygenase [EC:1.13.11.52] |
| IL-6 | interleukin-6 |
| KMO | kynureine 3-monooxygenase [EC:1.14.13.9] |
| KYNU | kynureninase [EC:3.7.1.3] |
| LDL | Low-density lipoprotein |
| MCP-1 | monocyte chemoattractant protein-1 |
| NA | nicotinic acid |
| NAAD | nicotinic acid adenine dinucleotide |
| NAD+ | nicotinamide adenine dinucleotide |
| NADK | NAD+ kinase [EC: 2.7.1.23] |
| NADP+ | nicotinamide adenine dinucleotide phosphate |
| NADS | glutamine-dependent NAD+ synthetase [EC:6.3.5.1] |
| NAM | nicotinamide |
| NAMN | nicotinic acid mononucleotide |
| NAMPT | nicotinamide phosphoribosyltransferase [EC:2.4.2.12] |
| NAPRT | nicotinate phosphoribosyltransferase [EC:6.3.4.21] |
| NAR | nicotinic acid riboside |
| NLRP3 | nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 |
| NMN | nicotinamide mononucleotide |
| NMNAT | nicotinamide mononucleotide adenylyltransferase [EC:2.7.7.1 2.7.7.18] |
| NNMT | nicotinamide N-methyltranserase [EC:2.1.1.1] |
| NO | nitric oxide |
| NR | nicotinamide ribose |
| NRK | nicotinamide riboside kinase [EC: 2.7.1.173] |
| PARP | poly-ADP-ribose polymerase [EC:2.4.2.30] |
| PBMC | peripheral blood mononuclear cells |
| PGE2 | prostaglandin E2 |
| Pi | phosphate |
| PPi | pyrophosphate |
| PRPP | phosphoribosyl pyrophosphate |
| QPRT | quinolate phosphoribosyl tranferase [EC:2.4.2.19] |
| RBCs | red blood cells |
| SAM | S-adenosyl-L-methionine |
| SAH | S-adenosyl-L-homocysteine |
| SEV | sevelamer |
| SIRT | sirtuin |
| TAG | triacylglycerol |
| TDO | tryptophan 2,3-dioxygenase [EC:1.13.11.11] |
| TGFβ1 | transforming growth factor beta 1 |
| TNFα | tumor necrosis factor alpha |
| UUO | unilateral ureteric obstruction |
| VCAM-1 | vascular cell adhesion protein-1 |
References
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ Metabolism: Pathophysiologic Mechanisms and Therapeutic Potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A.; Dam, A.; Maron, B.A.; Scribner, A.W.; Liao, R.; Handy, D.E.; Stanton, R.C.; Pitt, B.; Loscalzo, J. Aldosterone Impairs Vascular Reactivity by Decreasing Glucose-6-Phosphate Dehydrogenase Activity. Nat. Med. 2007, 13, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Poly(ADP-Ribose) Polymerase Inhibition: Past, Present and Future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Martin, D.R.; Lewington, A.J.; Hammerman, M.R.; Padanilam, B.J. Inhibition of Poly(ADP-Ribose) Polymerase Attenuates Ischemic Renal Injury in Rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1834–R1840. [Google Scholar] [CrossRef]
- Liu, S.; Liu, J.; Liu, D.; Wang, X.; Yang, R. Inhibition of Poly-(ADP-Ribose) Polymerase Protects the Kidney in a Canine Model of Endotoxic Shock. Nephron 2015, 130, 281–292. [Google Scholar] [CrossRef]
- Corda, D.; Di Girolamo, M. Functional Aspects of Protein Mono-ADP-Ribosylation. EMBO J. 2003, 22, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.-J.; Zhang, T.-N.; Chen, H.-H.; Yu, X.-F.; Lv, J.-L.; Liu, Y.-Y.; Liu, Y.-S.; Zheng, G.; Zhao, J.-Q.; Wei, Y.-F.; et al. The Sirtuin Family in Health and Disease. Signal Transduct. Target. Ther. 2022, 7, 402. [Google Scholar] [CrossRef]
- Braidy, N.; Berg, J.; Clement, J.; Khorshidi, F.; Poljak, A.; Jayasena, T.; Grant, R.; Sachdev, P. Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxid. Redox Signal. 2019, 30, 251–294. [Google Scholar] [CrossRef]
- Galione, A. Cyclic ADP-Ribose, the ADP-Ribosyl Cyclase Pathway and Calcium Signalling. Mol. Cell. Endocrinol. 1994, 98, 125–131. [Google Scholar] [CrossRef]
- Csiszar, A.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Kiss, T.; Farkas, E.; Baur, J.A.; Ungvari, Z. Role of Endothelial NAD+ Deficiency in Age-Related Vascular Dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1253–H1266. [Google Scholar] [CrossRef] [PubMed]
- Freeberg, K.A.; Udovich, C.C.; Martens, C.R.; Seals, D.R.; Craighead, D.H. Dietary Supplementation with NAD+-Boosting Compounds in Humans: Current Knowledge and Future Directions. J. Gerontol. Ser. A 2023, 78, 2435–2448. [Google Scholar] [CrossRef]
- Okabe, K.; Yaku, K.; Tobe, K.; Nakagawa, T. Implications of Altered NAD Metabolism in Metabolic Disorders. J. Biomed. Sci. 2019, 26, 34. [Google Scholar] [CrossRef] [PubMed]
- Campagna, R.; Vignini, A. NAD+ Homeostasis and NAD+-Consuming Enzymes: Implications for Vascular Health. Antioxidants 2023, 12, 376. [Google Scholar] [CrossRef] [PubMed]
- Felsted, R.L.; Chaykin, S. N1-Methylnicotinamide Oxidation in a Number of Mammals. J. Biol. Chem. 1967, 242, 1274–1279. [Google Scholar] [CrossRef]
- Rutkowski, B.; Slominska, E.; Szolkiewicz, M.; Smolenski, R.T.; Striley, C.; Rutkowski, P.; Swierczynski, J. N-Methyl-2-Pyridone-5-Carboxamide: A Novel Uremic Toxin? Kidney Int. 2003, 63, S19–S21. [Google Scholar] [CrossRef]
- Lenglet, A.; Liabeuf, S.; El Esper, N.; Brisset, S.; Mansour, J.; Lemaire-Hurtel, A.-S.; Mary, A.; Brazier, M.; Kamel, S.; Mentaverri, R.; et al. Efficacy and Safety of Nicotinamide in Haemodialysis Patients: The NICOREN Study. Nephrol. Dial. Transplant. 2017, 32, 870–879. [Google Scholar] [CrossRef]
- Ferrell, M.; Wang, Z.; Anderson, J.T.; Li, X.S.; Witkowski, M.; DiDonato, J.A.; Hilser, J.R.; Hartiala, J.A.; Haghikia, A.; Cajka, T.; et al. A Terminal Metabolite of Niacin Promotes Vascular Inflammation and Contributes to Cardiovascular Disease Risk. Nat. Med. 2024, 30, 424–434. [Google Scholar] [CrossRef]
- Kawai, S.; Murata, K. Structure and Function of NAD Kinase and NADP Phosphatase: Key Enzymes That Regulate the Intracellular Balance of NAD(H) and NADP(H). Biosci. Biotechnol. Biochem. 2008, 72, 919–930. [Google Scholar] [CrossRef]
- Liu, L.; Su, X.; Quinn, W.J.; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e5. [Google Scholar] [CrossRef]
- Çatak, J. Determination of Niacin Profiles in Some Animal and Plant Based Foods by High Performance Liquid Chromatography: Association with Healthy Nutrition. J. Anim. Sci. Technol. 2019, 61, 138–146. [Google Scholar] [CrossRef]
- Prabhu, D.; Dawe, R.S.; Mponda, K. Pellagra a Review Exploring Causes and Mechanisms, Including Isoniazid-Induced Pellagra. Photodermatol. Photoimmunol. Photomed. 2021, 37, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Andraos, S.; Jones, B.; Wall, C.; Thorstensen, E.; Kussmann, M.; Cameron-Smith, D.; Lange, K.; Clifford, S.; Saffery, R.; Burgner, D.; et al. Plasma B Vitamers: Population Epidemiology and Parent-Child Concordance in Children and Adults. Nutrients 2021, 13, 821. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD+ in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef]
- Ralto, K.M.; Rhee, E.P.; Parikh, S.M. NAD+ Homeostasis in Renal Health and Disease. Nat. Rev. Nephrol. 2020, 16, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.; Gille, A.; Zwykiel, S.; Lukasova, M.; Clausen, B.E.; Ahmed, K.; Tunaru, S.; Wirth, A.; Offermanns, S. Nicotinic Acid- and Monomethyl Fumarate-Induced Flushing Involves GPR109A Expressed by Keratinocytes and COX-2-Dependent Prostanoid Formation in Mice. J. Clin. Invest. 2010, 120, 2910–2919. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, X.; Bheda, P.; Revollo, J.R.; Imai, S.; Wolberger, C. Structure of Nampt/PBEF/Visfatin, a Mammalian NAD+ Biosynthetic Enzyme. Nat. Struct. Mol. Biol. 2006, 13, 661–662. [Google Scholar] [CrossRef]
- Shats, I.; Williams, J.G.; Liu, J.; Makarov, M.V.; Wu, X.; Lih, F.B.; Deterding, L.J.; Lim, C.; Xu, X.; Randall, T.A.; et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. 2020, 31, 564–579.e7. [Google Scholar] [CrossRef]
- Gardell, S.J.; Hopf, M.; Khan, A.; Dispagna, M.; Hampton Sessions, E.; Falter, R.; Kapoor, N.; Brooks, J.; Culver, J.; Petucci, C.; et al. Boosting NAD+ with a Small Molecule That Activates NAMPT. Nat. Commun. 2019, 10, 3241. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of Nicotinamide Riboside as a Nutrient and Conserved NRK Genes Establish a Preiss-Handler Independent Route to NAD+ in Fungi and Humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Mehmel, M.; Jovanović, N.; Spitz, U. Nicotinamide Riboside—The Current State of Research and Therapeutic Uses. Nutrients 2020, 12, 1616. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ Metabolism and Its Roles in Cellular Processes during Ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef]
- Mateuszuk, Ł.; Campagna, R.; Kutryb-Zając, B.; Kuś, K.; Słominska, E.M.; Smolenski, R.T.; Chlopicki, S. Reversal of Endothelial Dysfunction by Nicotinamide Mononucleotide via Extracellular Conversion to Nicotinamide Riboside. Biochem. Pharmacol. 2020, 178, 114019. [Google Scholar] [CrossRef] [PubMed]
- Beutelspacher, S.C.; Tan, P.H.; McClure, M.O.; Larkin, D.F.P.; Lechler, R.I.; George, A.J.T. Expression of Indoleamine 2,3-Dioxygenase (IDO) by Endothelial Cells: Implications for the Control of Alloresponses. Am. J. Transplant. 2006, 6, 1320–1330. [Google Scholar] [CrossRef]
- Shibata, K. Organ Co-Relationship in Tryptophan Metabolism and Factors That Govern the Biosynthesis of Nicotinamide from Tryptophan. J. Nutr. Sci. Vitaminol. 2018, 64, 90–98. [Google Scholar] [CrossRef]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De Novo NAD+ Synthesis Enhances Mitochondrial Function and Improves Health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef]
- Terakata, M.; Fukuwatari, T.; Sano, M.; Nakao, N.; Sasaki, R.; Fukuoka, S.-I.; Shibata, K. Establishment of True Niacin Deficiency in Quinolinic Acid Phosphoribosyltransferase Knockout Mice. J. Nutr. 2012, 142, 2148–2153. [Google Scholar] [CrossRef]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de Novo NAD+ Synthesis Specifies Immune Function in Aging and Inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- Pissios, P. Nicotinamide N-Methyltransferase: More Than a Vitamin B3 Clearance Enzyme. Trends Endocrinol. Metab. 2017, 28, 340–353. [Google Scholar] [CrossRef]
- Campagna, R.; Mazzanti, L.; Pompei, V.; Alia, S.; Vignini, A.; Emanuelli, M. The Multifaceted Role of Endothelial Sirt1 in Vascular Aging: An Update. Cells 2024, 13, 1469. [Google Scholar] [CrossRef] [PubMed]
- Isoherranen, N.; Thummel, K.E. Drug Metabolism and Transport during Pregnancy: How Does Drug Disposition Change during Pregnancy and What Are the Mechanisms That Cause Such Changes? Drug Metab. Dispos. 2013, 41, 256–262. [Google Scholar] [CrossRef]
- Heit, C.; Dong, H.; Chen, Y.; Thompson, D.C.; Deitrich, R.A.; Vasiliou, V.K. The Role of CYP2E1 in Alcohol Metabolism and Sensitivity in the Central Nervous System. In Cytochrome P450 2E1: Its Role in Disease and Drug Metabolism; Springer: Dordrecht, The Netherlands, 2013; Volume 67, pp. 235–247. [Google Scholar] [CrossRef]
- Menon, R.M.; Adams, M.H.; González, M.A.; Tolbert, D.S.; Leu, J.H.; Cefali, E.A. Plasma and Urine Pharmacokinetics of Niacin and Its Metabolites from an Extended-Release Niacin Formulation. Int. J. Clin. Pharmacol. Ther. 2007, 45, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Slominska, E.M.; Kowalik, K.; Smolenski, R.T.; Szolkiewicz, M.; Rutkowski, P.; Rutkowski, B.; Swierczynski, J. Accumulation of Poly(ADP-Ribose) Polymerase Inhibitors in Children with Chronic Renal Failure. Pediatr. Nephrol. 2006, 21, 800–806. [Google Scholar] [CrossRef]
- Rutkowski, B.; Swierczynski, J.; Slominska, E.; Szolkiewicz, M.; Smolenski, R.T.; Marlewski, M.; Butto, B.; Rutkowski, P. Disturbances of Purine Nucleotide Metabolism in Uremia. Semin. Nephrol. 2004, 24, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, P.; Malgorzewicz, S.; Slominska, E.; Renke, M.; Lysiak-Szydlowska, W.; Swierczynski, J.; Rutkowski, B. Interrelationship between Uremic Toxicity and Oxidative Stress. J. Ren. Nutr. 2006, 16, 190–193. [Google Scholar] [CrossRef]
- Rutkowski, P.; Słominska, E.M.; Szołkiewicz, M.; Aleksandrowicz, E.; Smolenski, R.T.; Wołyniec, W.; Renke, M.; Wisterowicz, K.; Swierczynski, J.; Rutkowski, B. Relationship between Uremic Toxins and Oxidative Stress in Patients with Chronic Renal Failure. Scand. J. Urol. Nephrol. 2007, 41, 243–248. [Google Scholar] [CrossRef]
- Slominska, E.M.; Smolenski, R.T.; Szolkiewicz, M.; Leaver, N.; Rutkowski, B.; Simmonds, H.A.; Swierczynski, J. Accumulation of Plasma N-Methyl-2-Pyridone-5-Carboxamide in Patients with Chronic Renal Failure. Mol. Cell. Biochem. 2002, 231, 83–88. [Google Scholar] [CrossRef]
- Lenglet, A.; Liabeuf, S.; Bodeau, S.; Louvet, L.; Mary, A.; Boullier, A.; Lemaire-Hurtel, A.S.; Jonet, A.; Sonnet, P.; Kamel, S.; et al. N-Methyl-2-Pyridone-5-Carboxamide (2PY)—Major Metabolite of Nicotinamide: An Update on an Old Uremic Toxin. Toxins 2016, 8, 339. [Google Scholar] [CrossRef]
- Jankowski, J.; Floege, J.; Fliser, D.; Böhm, M.; Marx, N. Cardiovascular Disease in Chronic Kidney Disease: Pathophysiological Insights and Therapeutic Options. Circulation 2021, 143, 1157–1172. [Google Scholar] [CrossRef]
- Pencina, K.M.; Valderrabano, R.; Wipper, B.; Orkaby, A.R.; Reid, K.F.; Storer, T.; Lin, A.P.; Merugumala, S.; Wilson, L.; Latham, N.; et al. Nicotinamide Adenine Dinucleotide Augmentation in Overweight or Obese Middle-Aged and Older Adults: A Physiologic Study. J. Clin. Endocrinol. Metab. 2023, 108, 1968–1980. [Google Scholar] [CrossRef]
- Yoshimura, N.; Yamada, K.; Ono, T.; Notoya, M.; Yukioka, H.; Takahashi, R.; Wakino, S.; Kanda, T.; Itoh, H. N-Methyl-2-Pyridone-5-Carboxamide (N-Me-2PY) Has Potent Anti-Fibrotic and Anti-Inflammatory Activity in a Fibrotic Kidney Model: Is It an Old Uremic Toxin? Clin. Exp. Nephrol. 2023, 27, 901–911. [Google Scholar] [CrossRef]
- Azuma, A. Pirfenidone Treatment of Idiopathic Pulmonary Fibrosis. Ther. Adv. Respir. Dis. 2012, 6, 107–114. [Google Scholar] [CrossRef] [PubMed]
- RamachandraRao, S.P.; Zhu, Y.; Ravasi, T.; McGowan, T.A.; Toh, I.; Dunn, S.R.; Okada, S.; Shaw, M.A.; Sharma, K. Pirfenidone Is Renoprotective in Diabetic Kidney Disease. J. Am. Soc. Nephrol. 2009, 20, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Moltrasio, C.; Romagnuolo, M.; Marzano, A.V. NLRP3 Inflammasome and NLRP3-Related Autoinflammatory Diseases: From Cryopyrin Function to Targeted Therapies. Front. Immunol. 2022, 13, 1007705. [Google Scholar] [CrossRef] [PubMed]
- Weiner, D.E.; Carpenter, M.A.; Levey, A.S.; Ivanova, A.; Cole, E.H.; Hunsicker, L.; Kasiske, B.L.; Kim, S.J.; Kusek, J.W.; Bostom, A.G. Kidney Function and Risk of Cardiovascular Disease and Mortality in Kidney Transplant Recipients: The FAVORIT Trial. Am. J. Transplant. 2012, 12, 2437–2445. [Google Scholar] [CrossRef]
- Rangaswami, J.; Mathew, R.O.; Parasuraman, R.; Tantisattamo, E.; Lubetzky, M.; Rao, S.; Yaqub, M.S.; Birdwell, K.A.; Bennett, W.; Dalal, P.; et al. Cardiovascular Disease in the Kidney Transplant Recipient: Epidemiology, Diagnosis and Management Strategies. Nephrol. Dial. Transplant. 2019, 34, 760–773. [Google Scholar] [CrossRef]
- Moist, L.M.; Port, F.K.; Orzol, S.M.; Young, E.W.; Ostbye, T.; Wolfe, R.A.; Hulbert-Shearon, T.; Jones, C.A.; Bloembergen, W.E. Predictors of Loss of Residual Renal Function among New Dialysis Patients. J. Am. Soc. Nephrol. 2000, 11, 556–564. [Google Scholar] [CrossRef]
- McIntyre, C.W. Recurrent Circulatory Stress: The Dark Side of Dialysis. Semin. Dial. 2010, 23, 449–451. [Google Scholar] [CrossRef]
- Cozzolino, M.; Mangano, M.; Stucchi, A.; Ciceri, P.; Conte, F.; Galassi, A. Cardiovascular Disease in Dialysis Patients. Nephrol. Dial. Transplant. 2018, 33, iii28–iii34. [Google Scholar] [CrossRef] [PubMed]
- van Haren, M.J.; Zhang, Y.; Thijssen, V.; Buijs, N.; Gao, Y.; Mateuszuk, L.; Fedak, F.A.; Kij, A.; Campagna, R.; Sartini, D.; et al. Macrocyclic Peptides as Allosteric Inhibitors of Nicotinamide N-Methyltransferase (NNMT). RSC Chem. Biol. 2021, 2, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; van Haren, M.J.; Buijs, N.; Innocenti, P.; Zhang, Y.; Sartini, D.; Campagna, R.; Emanuelli, M.; Parsons, R.B.; Jespers, W.; et al. Potent Inhibition of Nicotinamide N-Methyltransferase by Alkene-Linked Bisubstrate Mimics Bearing Electron Deficient Aromatics. J. Med. Chem. 2021, 64, 12938–12963. [Google Scholar] [CrossRef]
- van Haren, M.J.; Gao, Y.; Buijs, N.; Campagna, R.; Sartini, D.; Emanuelli, M.; Mateuszuk, L.; Kij, A.; Chlopicki, S.; Escudé Martinez de Castilla, P.; et al. Esterase-Sensitive Prodrugs of a Potent Bisubstrate Inhibitor of Nicotinamide N-Methyltransferase (NNMT) Display Cellular Activity. Biomolecules 2021, 11, 1357. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M. Supplementation with NAD+ and Its Precursors to Prevent Cognitive Decline across Disease Contexts. Nutrients 2022, 14, 3231. [Google Scholar] [CrossRef]
- Feng, Z.; Qin, Y.; Huo, F.; Jian, Z.; Li, X.; Geng, J.; Li, Y.; Wu, J. NMN Recruits GSH to Enhance GPX4-Mediated Ferroptosis Defense in UV Irradiation Induced Skin Injury. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2022, 1868, 166287. [Google Scholar] [CrossRef]
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of Silencing and Accelerated Aging by Nicotinamide, a Putative Negative Regulator of Yeast Sir2 and Human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. [Google Scholar] [CrossRef]
- Yoshino, M.; Yoshino, J.; Kayser, B.D.; Patti, G.J.; Franczyk, M.P.; Mills, K.F.; Sindelar, M.; Pietka, T.; Patterson, B.W.; Imai, S.-I.; et al. Nicotinamide Mononucleotide Increases Muscle Insulin Sensitivity in Prediabetic Women. Science 2021, 372, 1224–1229. [Google Scholar] [CrossRef]
- Yi, L.; Maier, A.B.; Tao, R.; Lin, Z.; Vaidya, A.; Pendse, S.; Thasma, S.; Andhalkar, N.; Avhad, G.; Kumbhar, V. The Efficacy and Safety of β-Nicotinamide Mononucleotide (NMN) Supplementation in Healthy Middle-Aged Adults: A Randomized, Multicenter, Double-Blind, Placebo-Controlled, Parallel-Group, Dose-Dependent Clinical Trial. GeroScience 2023, 45, 29–43. [Google Scholar] [CrossRef]
- Song, Q.; Zhou, X.; Xu, K.; Liu, S.; Zhu, X.; Yang, J. The Safety and Antiaging Effects of Nicotinamide Mononucleotide in Human Clinical Trials: An Update. Adv. Nutr. 2023, 14, 1416–1435. [Google Scholar] [CrossRef]
- Elhassan, Y.S.; Kluckova, K.; Fletcher, R.S.; Schmidt, M.S.; Garten, A.; Doig, C.L.; Cartwright, D.M.; Oakey, L.; Burley, C.V.; Jenkinson, N.; et al. Nicotinamide Riboside Augments the Aged Human Skeletal Muscle NAD+ Metabolome and Induces Transcriptomic and Anti-Inflammatory Signatures. Cell Rep. 2019, 28, 1717–1728.e6. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide Riboside Is Uniquely and Orally Bioavailable in Mice and Humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef] [PubMed]
- Veenhuis, S.J.G.; van Os, N.J.H.; Janssen, A.J.W.M.; van Gerven, M.H.J.C.; Coene, K.L.M.; Engelke, U.F.H.; Wevers, R.A.; Tinnevelt, G.H.; Ter Heine, R.; van de Warrenburg, B.P.C.; et al. Nicotinamide Riboside Improves Ataxia Scores and Immunoglobulin Levels in Ataxia Telangiectasia. Mov. Disord. 2021, 36, 2951–2957. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Douek, I.F.; Moore, W.P.; Gillmor, H.A.; McLean, A.E.; Bingley, P.J.; Gale, E.A. European Nicotinamide Diabetes Intervention Trial Group Safety of High-Dose Nicotinamide: A Review. Diabetologia 2000, 43, 1337–1345. [Google Scholar] [CrossRef]
- Fischer, L.J.; Falany, J.; Fisher, R. Characteristics of Nicotinamide and N1-Methylnicotinamide Protection from Alloxan Diabetes in Mice. Toxicol. Appl. Pharmacol. 1983, 70, 148–155. [Google Scholar] [CrossRef]
- Biedroń, R.; Ciszek, M.; Tokarczyk, M.; Bobek, M.; Kurnyta, M.; Słominska, E.M.; Smoleński, R.T.; Marcinkiewicz, J. 1-Methylnicotinamide and Nicotinamide: Two Related Anti-Inflammatory Agents That Differentially Affect the Functions of Activated Macrophages. Arch. Immunol. Ther. Exp. 2008, 56, 127–134. [Google Scholar] [CrossRef]
- Goldsmith, D.R.; Scott, L.J.; Cvetković, R.S.; Plosker, G.L. Sevelamer Hydrochloride: A Review of Its Use for Hyperphosphataemia in Patients with End-Stage Renal Disease on Haemodialysis. Drugs 2008, 68, 85–104. [Google Scholar] [CrossRef]
- O’Connell, C.; Horwood, K.; Nadamuni, M. Correction of Refractory Thrombocytopenia and Anemia Following Withdrawal of Extended Release Niacin. Am. J. Hematol. 2016, 91, E318. [Google Scholar] [CrossRef]
- Chlopicki, S.; Swies, J.; Mogielnicki, A.; Buczko, W.; Bartus, M.; Lomnicka, M.; Adamus, J.; Gebicki, J. 1-Methylnicotinamide (MNA), a Primary Metabolite of Nicotinamide, Exerts Anti-Thrombotic Activity Mediated by a Cyclooxygenase-2/Prostacyclin Pathway. Br. J. Pharmacol. 2007, 152, 230–239. [Google Scholar] [CrossRef]
- Bartuś, M.; Łomnicka, M.; Kostogrys, R.B.; Kaźmierczak, P.; Watała, C.; Słominska, E.M.; Smoleński, R.T.; Pisulewski, P.M.; Adamus, J.; Gebicki, J.; et al. 1-Methylnicotinamide (MNA) Prevents Endothelial Dysfunction in Hypertriglyceridemic and Diabetic Rats. Pharmacol. Rep. 2008, 60, 127–138. [Google Scholar]
- Watała, C.; Kaźmierczak, P.; Dobaczewski, M.; Przygodzki, T.; Bartuś, M.; Łomnicka, M.; Słomińska, E.M.; Durackova, Z.; Chłopicki, S. Anti-Diabetic Effects of 1-Methylnicotinamide (MNA) in Streptozocin-Induced Diabetes in Rats. Pharmacol. Rep. 2009, 61, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Brzozowski, T.; Konturek, P.C.; Chlopicki, S.; Sliwowski, Z.; Pawlik, M.; Ptak-Belowska, A.; Kwiecień, S.; Drozdowicz, D.; Pajdo, R.; Slonimska, E.; et al. Therapeutic Potential of 1-Methylnicotinamide against Acute Gastric Lesions Induced by Stress: Role of Endogenous Prostacyclin and Sensory Nerves. J. Pharmacol. Exp. Ther. 2008, 326, 105–116. [Google Scholar] [CrossRef]
- Camillo, L.; Zavattaro, E.; Savoia, P. Nicotinamide: A Multifaceted Molecule in Skin Health and Beyond. Medicina 2025, 61, 254. [Google Scholar] [CrossRef] [PubMed]
- Zackheim, H.S. Topical 6-Aminonicotinamide Plus Oral Niacinamide Therapy for Psoriasis. Arch. Dermatol. 1978, 114, 1632–1638. [Google Scholar] [CrossRef]
- Wozniacka, A.; Wieczorkowska, M.; Gebicki, J.; Sysa-Jedrzejowska, A. Topical Application of 1-Methylnicotinamide in the Treatment of Rosacea: A Pilot Study. Clin. Exp. Dermatol. 2005, 30, 632–635. [Google Scholar] [CrossRef] [PubMed]
- Pumpo, R.; Sarnelli, G.; Spinella, A.; Budillon, G.; Cuomo, R. The Metabolism of Nicotinamide in Human Liver Cirrhosis: A Study on N-Methylnicotinamide and 2-Pyridone-5-Carboxamide Production. Am. J. Gastroenterol. 2001, 96, 1183–1187. [Google Scholar] [CrossRef]
- Cazzullo, C.L.; Sacchetti, E.; Smeraldi, E. N-Methylnicotinamide Excretion and Affective Disorders. Psychol. Med. 1976, 6, 265–270. [Google Scholar] [CrossRef]
- Aoyama, K.; Matsubara, K.; Okada, K.; Fukushima, S.; Shimizu, K.; Yamaguchi, S.; Uezono, T.; Satomi, M.; Hayase, N.; Ohta, S.; et al. N-Methylation Ability for Azaheterocyclic Amines Is Higher in Parkinson’s Disease: Nicotinamide Loading Test. J. Neural Transm. 2000, 107, 985–995. [Google Scholar] [CrossRef]
- Barlow, G.B.; Sutton, J.L.; Wilkinson, A.W. Metabolism of Nicotinic Acid in Children with Burns and Scalds. Clin. Chim. Acta 1977, 75, 337–342. [Google Scholar] [CrossRef]
- Altschul, R.; Hoffer, A.; Stephen, J.D. Influence of Nicotinic Acid on Serum Cholesterol in Man. Arch. Biochem. Biophys. 1955, 54, 558–559. [Google Scholar] [CrossRef]
- Bruckert, E.; Labreuche, J.; Amarenco, P. Meta-Analysis of the Effect of Nicotinic Acid Alone or in Combination on Cardiovascular Events and Atherosclerosis. Atherosclerosis 2010, 210, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Duggal, J.K.; Singh, M.; Attri, N.; Singh, P.P.; Ahmed, N.; Pahwa, S.; Molnar, J.; Singh, S.; Khosla, S.; Arora, R. Effect of Niacin Therapy on Cardiovascular Outcomes in Patients with Coronary Artery Disease. J. Cardiovasc. Pharmacol. Ther. 2010, 15, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J. Clofibrate and Niacin in Coronary Heart Disease. JAMA 1975, 231, 360–381. [Google Scholar] [CrossRef]
- Kamanna, V.S.; Kashyap, M.L. Mechanism of Action of Niacin on Lipoprotein Metabolism. Curr. Atheroscler. Rep. 2000, 2, 36–46. [Google Scholar] [CrossRef]
- Digby, J.E.; Ruparelia, N.; Choudhury, R.P. Niacin in Cardiovascular Disease: Recent Preclinical and Clinical Developments. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 582–588. [Google Scholar] [CrossRef]
- Lavigne, P.M.; Karas, R.H. The Current State of Niacin in Cardiovascular Disease Prevention: A Systematic Review and Meta-Regression. J. Am. Coll. Cardiol. 2013, 61, 440–446. [Google Scholar] [CrossRef]
- Berrougui, H.; Momo, C.N.; Khalil, A. Health Benefits of High-Density Lipoproteins in Preventing Cardiovascular Diseases. J. Clin. Lipidol. 2012, 6, 524–533. [Google Scholar] [CrossRef]
- Wu, B.J.; Yan, L.; Charlton, F.; Witting, P.; Barter, P.J.; Rye, K.-A. Evidence That Niacin Inhibits Acute Vascular Inflammation and Improves Endothelial Dysfunction Independent of Changes in Plasma Lipids. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 968–975. [Google Scholar] [CrossRef]
- Plaisance, E.P.; Lukasova, M.; Offermanns, S.; Zhang, Y.; Cao, G.; Judd, R.L. Niacin Stimulates Adiponectin Secretion through the GPR109A Receptor. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E549–E558. [Google Scholar] [CrossRef]
- Ouchi, N.; Kihara, S.; Funahashi, T.; Matsuzawa, Y.; Walsh, K. Obesity, Adiponectin and Vascular Inflammatory Disease. Curr. Opin. Lipidol. 2003, 14, 561–566. [Google Scholar] [CrossRef]
- Ganji, S.H.; Qin, S.; Zhang, L.; Kamanna, V.S.; Kashyap, M.L. Niacin Inhibits Vascular Oxidative Stress, Redox-Sensitive Genes, and Monocyte Adhesion to Human Aortic Endothelial Cells. Atherosclerosis 2009, 202, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Brown, B.G.; Zhao, X.Q.; Chait, A.; Fisher, L.D.; Cheung, M.C.; Morse, J.S.; Dowdy, A.A.; Marino, E.K.; Bolson, E.L.; Alaupovic, P.; et al. Simvastatin and Niacin, Antioxidant Vitamins, or the Combination for the Prevention of Coronary Disease. N. Engl. J. Med. 2001, 345, 1583–1592. [Google Scholar] [CrossRef]
- Taylor, A.J.; Sullenberger, L.E.; Lee, H.J.; Lee, J.K.; Grace, K.A. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: A Double-Blind, Placebo-Controlled Study of Extended-Release Niacin on Atherosclerosis Progression in Secondary Prevention Patients Treated with Statins. Circulation 2004, 110, 3512–3517. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.J.; Lee, H.J.; Sullenberger, L.E. The Effect of 24 Months of Combination Statin and Extended-Release Niacin on Carotid Intima-Media Thickness: ARBITER 3. Curr. Med. Res. Opin. 2006, 22, 2243–2250. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.J.; Villines, T.C.; Stanek, E.J.; Devine, P.J.; Griffen, L.; Miller, M.; Weissman, N.J.; Turco, M. Extended-Release Niacin or Ezetimibe and Carotid Intima-Media Thickness. N. Engl. J. Med. 2009, 361, 2113–2122. [Google Scholar] [CrossRef]
- Lee, J.M.S.; Robson, M.D.; Yu, L.-M.; Shirodaria, C.C.; Cunnington, C.; Kylintireas, I.; Digby, J.E.; Bannister, T.; Handa, A.; Wiesmann, F.; et al. Effects of High-Dose Modified-Release Nicotinic Acid on Atherosclerosis and Vascular Function: A Randomized, Placebo-Controlled, Magnetic Resonance Imaging Study. J. Am. Coll. Cardiol. 2009, 54, 1787–1794. [Google Scholar] [CrossRef]
- Villines, T.C.; Stanek, E.J.; Devine, P.J.; Turco, M.; Miller, M.; Weissman, N.J.; Griffen, L.; Taylor, A.J. The ARBITER 6-HALTS Trial (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol 6-HDL and LDL Treatment Strategies in Atherosclerosis): Final Results and the Impact of Medication Adherence, Dose, and Treatment Duration. J. Am. Coll. Cardiol. 2010, 55, 2721–2726. [Google Scholar] [CrossRef]
- Garg, A.; Sharma, A.; Krishnamoorthy, P.; Garg, J.; Virmani, D.; Sharma, T.; Stefanini, G.; Kostis, J.B.; Mukherjee, D.; Sikorskaya, E. Role of Niacin in Current Clinical Practice: A Systematic Review. Am. J. Med. 2017, 130, 173–187. [Google Scholar] [CrossRef]
- Makarov, M.V.; Trammell, S.A.J.; Migaud, M.E. The Chemistry of the Vitamin B3 Metabolome. Biochem. Soc. Trans. 2019, 47, 131–147. [Google Scholar] [CrossRef]
- Abelson, D.; Boyle, A.; Seligson, H. Identification of N’-Methyl-4-Pyridone-3-Carboxamide in Human Plasma. J. Biol. Chem. 1963, 238, 717–718. [Google Scholar] [CrossRef]
- Menon, R.M.; González, M.A.; Adams, M.H.; Tolbert, D.S.; Leu, J.H.; Cefali, E.A. Effect of the Rate of Niacin Administration on the Plasma and Urine Pharmacokinetics of Niacin and Its Metabolites. J. Clin. Pharmacol. 2007, 47, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A. Effect of Niacin on Endothelial Function: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Vasc. Med. Lond. Engl. 2014, 19, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Schandelmaier, S.; Briel, M.; Saccilotto, R.; Olu, K.K.; Arpagaus, A.; Hemkens, L.G.; Nordmann, A.J. Niacin for Primary and Secondary Prevention of Cardiovascular Events. Cochrane Database Syst. Rev. 2017, 6, CD009744. [Google Scholar] [CrossRef] [PubMed]
- He, Y.-M.; Feng, L.; Huo, D.-M.; Yang, Z.-H.; Liao, Y.-H. Benefits and Harm of Niacin and Its Analog for Renal Dialysis Patients: A Systematic Review and Meta-Analysis. Int. Urol. Nephrol. 2014, 46, 433–442. [Google Scholar] [CrossRef]
- EFSA. Nicotinic Acid and Nicotinamide for All Animal Species. Available online: https://www.efsa.europa.eu/en/efsajournal/pub/2781 (accessed on 26 February 2025).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dettlaff-Pokora, A.; Swierczynski, J. High Concentrations of Circulating 2PY and 4PY—Potential Risk Factor of Cardiovascular Disease in Patients with Chronic Kidney Disease. Int. J. Mol. Sci. 2025, 26, 4463. https://doi.org/10.3390/ijms26094463
Dettlaff-Pokora A, Swierczynski J. High Concentrations of Circulating 2PY and 4PY—Potential Risk Factor of Cardiovascular Disease in Patients with Chronic Kidney Disease. International Journal of Molecular Sciences. 2025; 26(9):4463. https://doi.org/10.3390/ijms26094463
Chicago/Turabian StyleDettlaff-Pokora, Agnieszka, and Julian Swierczynski. 2025. "High Concentrations of Circulating 2PY and 4PY—Potential Risk Factor of Cardiovascular Disease in Patients with Chronic Kidney Disease" International Journal of Molecular Sciences 26, no. 9: 4463. https://doi.org/10.3390/ijms26094463
APA StyleDettlaff-Pokora, A., & Swierczynski, J. (2025). High Concentrations of Circulating 2PY and 4PY—Potential Risk Factor of Cardiovascular Disease in Patients with Chronic Kidney Disease. International Journal of Molecular Sciences, 26(9), 4463. https://doi.org/10.3390/ijms26094463