
Mitochondrial Dysfunction in Genetic and Non-Genetic Parkinson’s Disease
 , ,
, ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
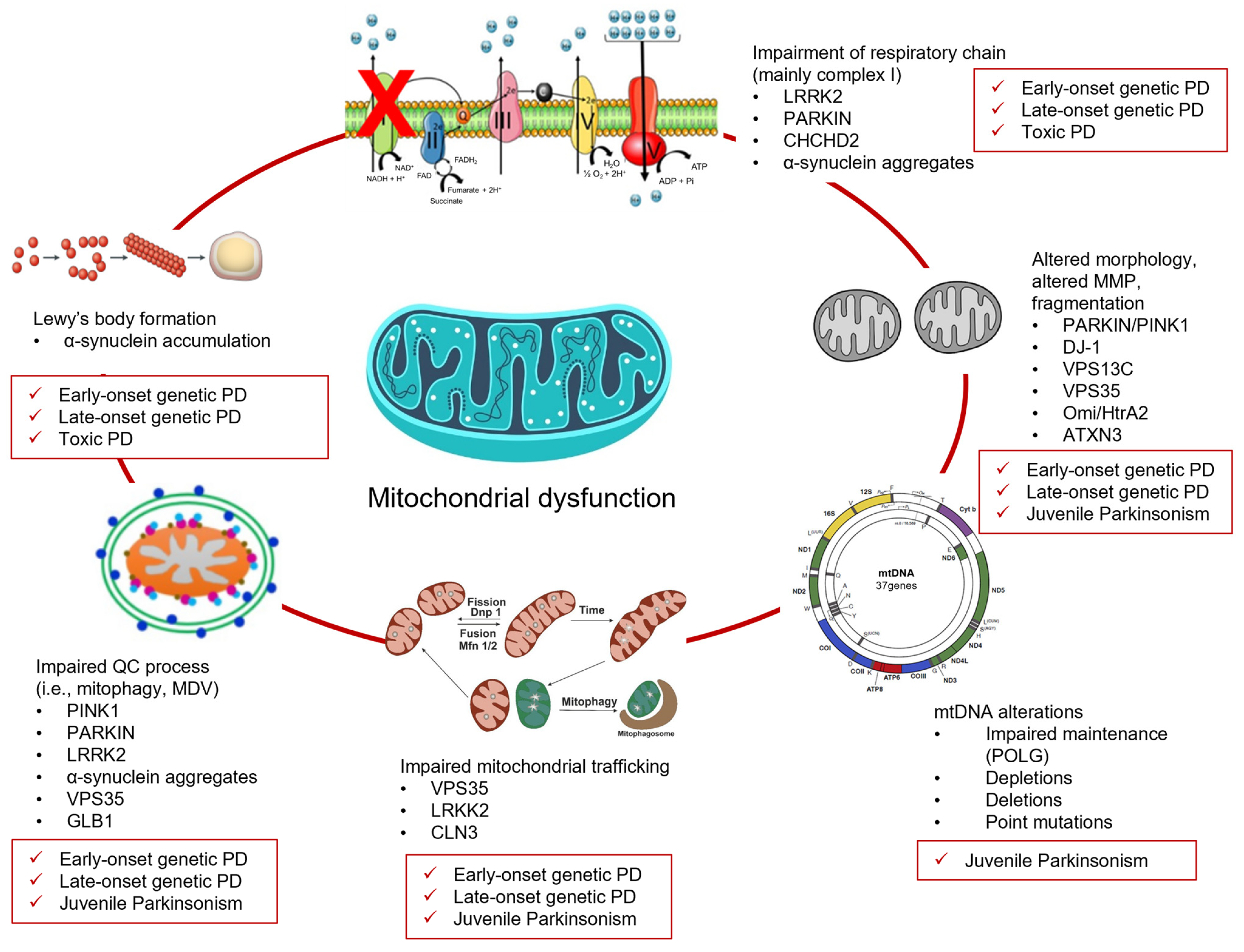
2. Juvenile- and Early-Onset Genetic PD and Mitochondria Dysfunction
2.1. Parkin (PARK2)
2.2. PINK1 (PARK6)
2.3. DJ-1 (PARK7)
2.4. ATP13A2 (PARK9)
2.5. PLA2G6 (PARK14)
2.6. FBXO7 (PARK15)
2.7. Vacuolar Protein Sorting 13C (VPS13C) (PARK23)
3. Late-Onset Genetic PD and Mitochondrial Dysfunction
3.1. SNCA (PARK1/PARK4)
3.2. LRRK2 (PARK8)
3.3. Omi/HtrA2 (PARK 13)
3.4. Vacuolar Protein Sorting 35 (VPS35) (PARK17)
3.5. Coiled-Helix-Coiled-Helix Domain Containing 2 (CHCHD2) (PARK22)
4. Juvenile Genetic Atypical Parkinsonian Syndromes and Mitochondrial Dysfunction
4.1. ATXN3
4.2. CLN3
4.3. GLB1
4.4. POLG
4.5. Hereditary Spastic Paraplegia (HSP)
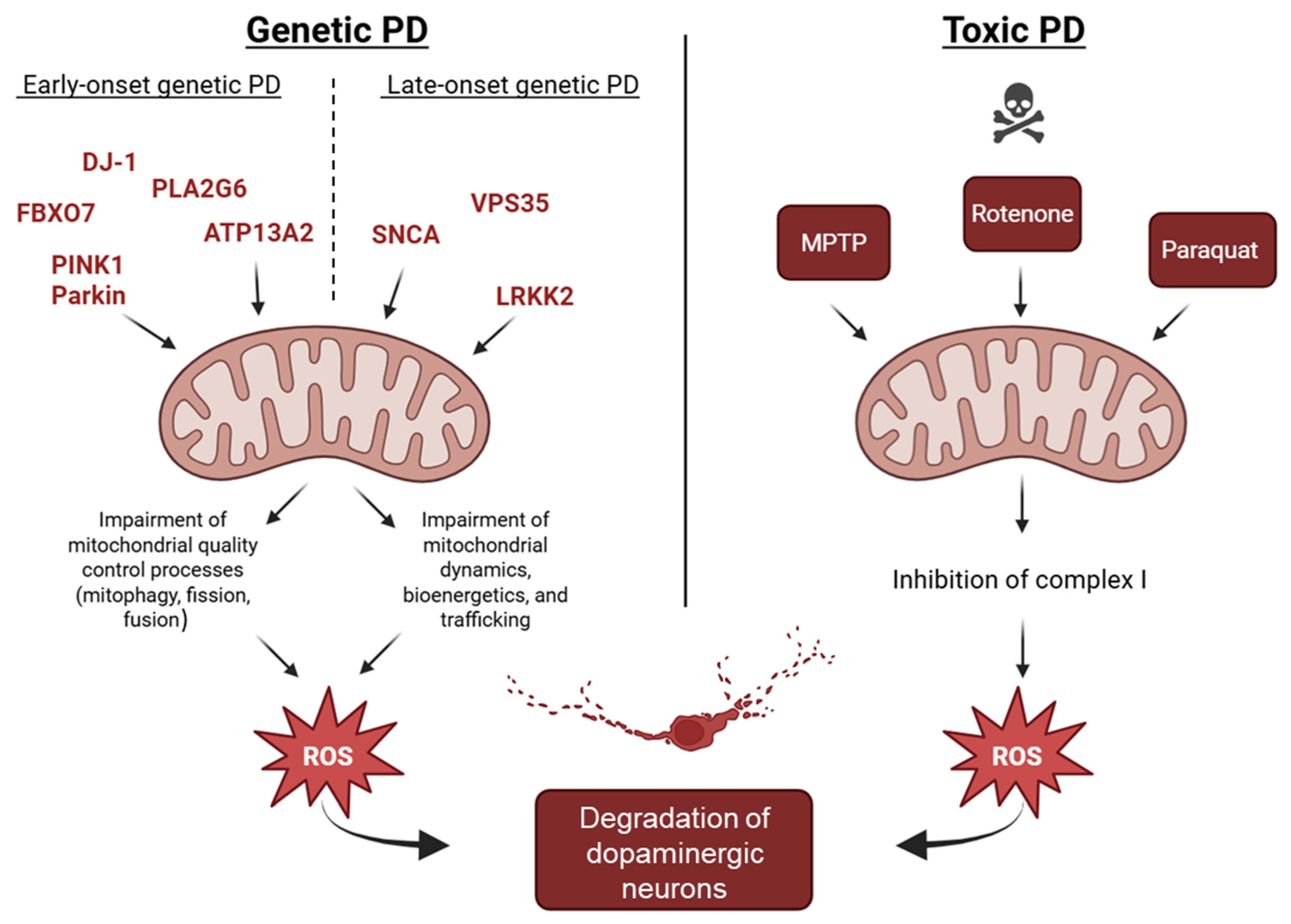
5. Environmental Toxins, PD, and Mitochondrial Dysfunction
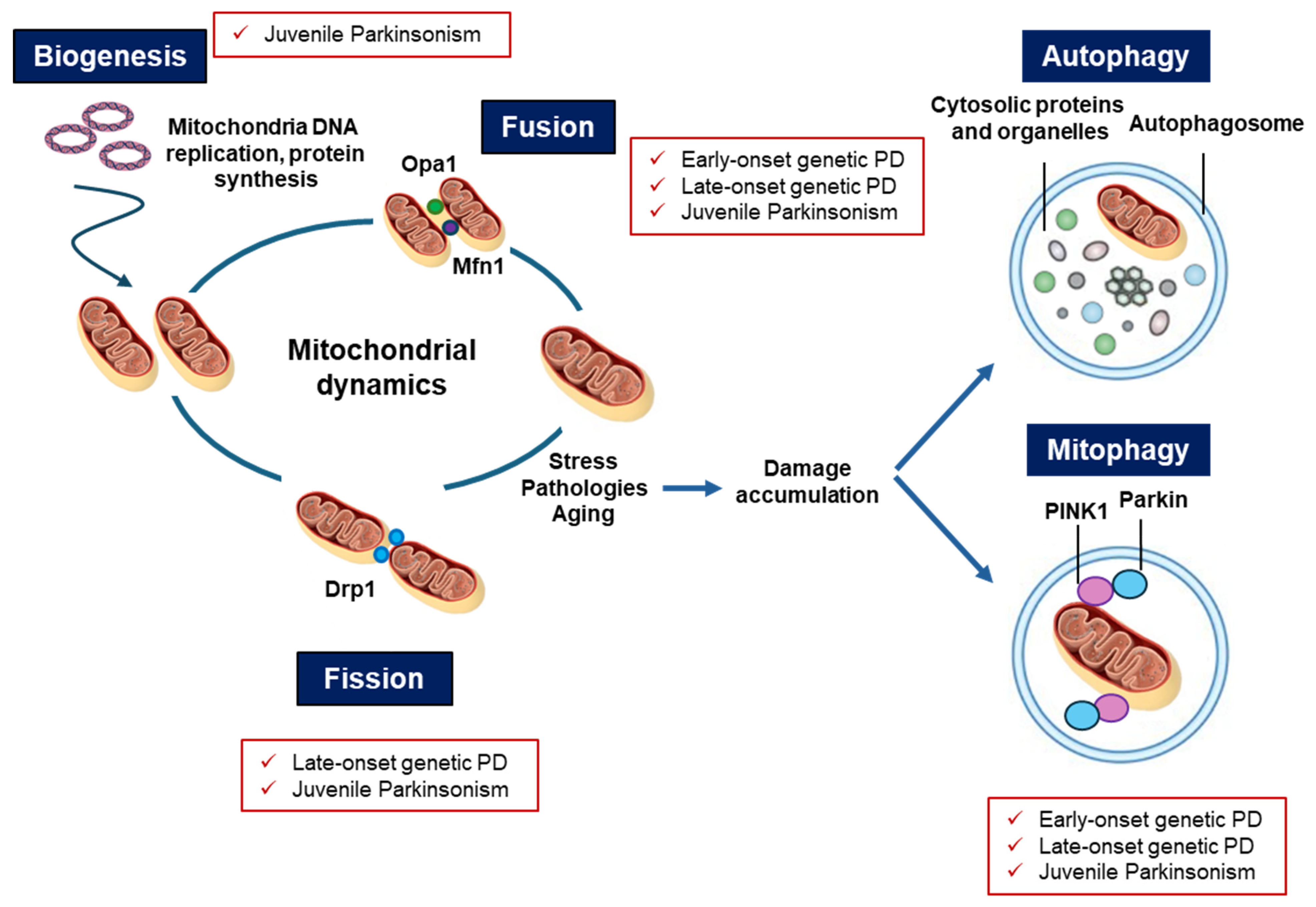
6. Converging Mechanisms in Parkinson’s Disease: The Mitochondrial Link Between Genetic and Non-Genetic Cases
7. Mitochondria-Targeted Therapeutic Approaches in PD
8. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Todorova, A.; Jenner, P.; Ray Chaudhuri, K. Non-Motor Parkinson’s: Integral to Motor Parkinson’s, yet Often Neglected. Pract. Neurol. 2014, 14, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Dolgacheva, L.P.; Zinchenko, V.P.; Goncharov, N.V. Molecular and Cellular Interactions in Pathogenesis of Sporadic Parkinson Disease. Int. J. Mol. Sci. 2022, 23, 13043. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Wang, P.; Jankovic, J. The Genetics of Parkinson Disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Lim, K.-L. Genetic Insights into Sporadic Parkinson’s Disease Pathogenesis. Curr. Genom. 2014, 14, 486–501. [Google Scholar] [CrossRef]
- das Chagas Campêlo, C.L.; Silva, R.H. Genetic Variants in SNCA and the Risk of Sporadic Parkinson’s Disease and Clinical Outcomes: A Review. Park. Dis. 2017, 2017, 4318416. [Google Scholar] [CrossRef]
- Belenguer, P.; Duarte, J.M.N.; Schuck, P.F.; Ferreira, G.C. Mitochondria and the Brain: Bioenergetics and Beyond. Neurotox. Res. 2019, 36, 219–238. [Google Scholar] [CrossRef]
- Tábara, L.-C.; Segawa, M.; Prudent, J. Molecular Mechanisms of Mitochondrial Dynamics. Nat. Rev. Mol. Cell Biol. 2025, 26, 123–146. [Google Scholar] [CrossRef]
- Chen, R.; Gu, X.; Wang, X. α-Synuclein in Parkinson’s Disease and Advances in Detection. Clin. Chim. Acta 2022, 529, 76–86. [Google Scholar] [CrossRef]
- Lin, K.-J.; Lin, K.-L.; Chen, S.-D.; Liou, C.-W.; Chuang, Y.-C.; Lin, H.-Y.; Lin, T.-K. The Overcrowded Crossroads: Mitochondria, Alpha-Synuclein, and the Endo-Lysosomal System Interaction in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef]
- Henrich, M.T.; Oertel, W.H.; Surmeier, D.J.; Geibl, F.F. Mitochondrial Dysfunction in Parkinson’s Disease—A Key Disease Hallmark with Therapeutic Potential. Mol. Neurodegener. 2023, 18, 83. [Google Scholar] [CrossRef]
- Scarselli, M.; Armogida, M.; Chiacchio, S.; DeMontis, M.G.; Colzi, A.; Corsini, G.U.; Maggio, R. Reconstitution of Functional Dopamine D2s Receptor by Co-Expression of Amino- and Carboxyl-Terminal Receptor Fragments. Eur. J. Pharmacol. 2000, 397, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The Genetic Architecture of Parkinson’s Disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; Pereira, S.L.; Klein, C.; Grünewald, A. Mitochondria and Parkinson’s Disease: Clinical, Molecular, and Translational Aspects. J. Park. Dis. 2021, 11, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s Disease. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial Import and Accumulation of α-Synuclein Impair Complex I in Human Dopaminergic Neuronal Cultures and Parkinson Disease Brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Flores-Ponce, X.; Velasco, I. Dopaminergic Neuron Metabolism: Relevance for Understanding Parkinson’s Disease. Metabolomics 2024, 20, 116. [Google Scholar] [CrossRef]
- Harraz, M.M. Selective Dopaminergic Vulnerability in Parkinson’s Disease: New Insights into the Role of DAT. Front. Neurosci. 2023, 17, 1219441. [Google Scholar] [CrossRef]
- Bourdenx, M.; Dehay, B. What Lysosomes Actually Tell Us about Parkinson’s Disease? Ageing Res. Rev. 2016, 32, 140–149. [Google Scholar] [CrossRef]
- Petese, A.; Cesaroni, V.; Cerri, S.; Blandini, F. Are Lysosomes Potential Therapeutic Targets for Parkinson’s Disease? CNS Neurol. Disord. Drug Targets 2022, 21, 642–655. [Google Scholar] [CrossRef]
- Smith, L.; Schapira, A.H.V. GBA Variants and Parkinson Disease: Mechanisms and Treatments. Cells 2022, 11, 1261. [Google Scholar] [CrossRef]
- Paviour, D.C.; Surtees, R.A.H.; Lees, A.J. Diagnostic Considerations in Juvenile Parkinsonism. Mov. Disord. 2004, 19, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Dogu, O.; Johnson, J.; Hernandez, D.; Hanson, M.; Hardy, J.; Apaydin, H.; Özekmekçi, S.; Sevim, S.; Gwinn-Hardy, K.; Singleton, A. A Consanguineous Turkish Family with Early-onset Parkinson’s Disease and an Exon 4 Parkin Deletion. Mov. Disord. 2004, 19, 812–816. [Google Scholar] [CrossRef] [PubMed]
- Olszewska, D.A.; McCarthy, A.; Soto-Beasley, A.I.; Walton, R.L.; Ross, O.A.; Lynch, T. PARKIN, PINK1, and DJ1 Analysis in Early-Onset Parkinson’s Disease in Ireland. Ir. J. Med. Sci. 2022, 191, 901–907. [Google Scholar] [CrossRef] [PubMed]
- Kolicheski, A.; Turcano, P.; Tamvaka, N.; McLean, P.J.; Springer, W.; Savica, R.; Ross, O.A. Early-Onset Parkinson’s Disease: Creating the Right Environment for a Genetic Disorder. J. Park. Dis. 2022, 12, 2353–2367. [Google Scholar] [CrossRef]
- Selikhova, M.; Williams, D.R.; Kempster, P.A.; Holton, J.L.; Revesz, T.; Lees, A.J. A Clinico-Pathological Study of Subtypes in Parkinson’s Disease. Brain 2009, 132, 2947–2957. [Google Scholar] [CrossRef]
- Ferguson, L.W.; Rajput, A.H.; Rajput, A. Early-Onset vs. Late-Onset Parkinson’s Disease: A Clinical-Pathological Study. Can. J. Neurol. Sci. 2016, 43, 113–119. [Google Scholar] [CrossRef]
- Mortiboys, H.; Thomas, K.J.; Koopman, W.J.H.; Klaffke, S.; Abou-Sleiman, P.; Olpin, S.; Wood, N.W.; Willems, P.H.G.M.; Smeitink, J.A.M.; Cookson, M.R.; et al. Mitochondrial Function and Morphology Are Impaired in Parkin-Mutant Fibroblasts. Ann. Neurol. 2008, 64, 555–565. [Google Scholar] [CrossRef]
- Flinn, L.J.; Keatinge, M.; Bretaud, S.; Mortiboys, H.; Matsui, H.; De Felice, E.; Woodroof, H.I.; Brown, L.; McTighe, A.; Soellner, R.; et al. TigarB Causes Mitochondrial Dysfunction and Neuronal Loss in PINK1 Deficiency. Ann. Neurol. 2013, 74, 837–847. [Google Scholar] [CrossRef]
- Flinn, L.; Mortiboys, H.; Volkmann, K.; Köster, R.W.; Ingham, P.W.; Bandmann, O. Complex I Deficiency and Dopaminergic Neuronal Cell Loss in Parkin-Deficient Zebrafish (Danio Rerio). Brain 2009, 132, 1613–1623. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 Is Selectively Stabilized on Impaired Mitochondria to Activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Lazarou, M.; Wang, C.; Kane, L.A.; Narendra, D.P.; Youle, R.J. Mitochondrial Membrane Potential Regulates PINK1 Import and Proteolytic Destabilization by PARL. J. Cell Biol. 2010, 191, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 Causes Mitochondrial Functional Defects and Increased Sensitivity to Oxidative Stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.-I.; Bower, A.; Jiang, H.; Kang, S.-U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin Loss Leads to PARIS-Dependent Declines in Mitochondrial Mass and Respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef]
- Haylett, W.; Swart, C.; van der Westhuizen, F.; van Dyk, H.; van der Merwe, L.; van der Merwe, C.; Loos, B.; Carr, J.; Kinnear, C.; Bardien, S. Altered Mitochondrial Respiration and Other Features of Mitochondrial Function in Parkin-Mutant Fibroblasts from Parkinson’s Disease Patients. Park. Dis. 2016, 2016, 819209. [Google Scholar] [CrossRef]
- Stauch, K.L.; Villeneuve, L.M.; Purnell, P.R.; Ottemann, B.M.; Emanuel, K.; Fox, H.S. Loss of Pink1 Modulates Synaptic Mitochondrial Bioenergetics in the Rat Striatum Prior to Motor Symptoms: Concomitant Complex I Respiratory Defects and Increased Complex II-Mediated Respiration. Proteom. Clin. Appl. 2016, 10, 1205–1217. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.J.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 Gene Associated with Autosomal Recessive Early-Onset Parkinsonism. Science (1979) 2003, 299, 256–259. [Google Scholar] [CrossRef]
- van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a Common Biological Pathway Linking Three Parkinson’s Disease-causing Genes: Parkin, PINK1 and DJ-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef]
- Blackinton, J.; Lakshminarasimhan, M.; Thomas, K.J.; Ahmad, R.; Greggio, E.; Raza, A.S.; Cookson, M.R.; Wilson, M.A. Formation of a Stabilized Cysteine Sulfinic Acid Is Critical for the Mitochondrial Function of the Parkinsonism Protein DJ-1. J. Biol. Chem. 2009, 284, 6476–6485. [Google Scholar] [CrossRef]
- Heo, J.Y.; Park, J.H.; Kim, S.J.; Seo, K.S.; Han, J.S.; Lee, S.H.; Kim, J.M.; Park, J., II; Park, S.K.; Lim, K.; et al. DJ-1 Null Dopaminergic Neuronal Cells Exhibit Defects in Mitochondrial Function and Structure: Involvement of Mitochondrial Complex I Assembly. PLoS ONE 2012, 7, e32629. [Google Scholar] [CrossRef]
- Trinh, J.; Farrer, M. Advances in the Genetics of Parkinson Disease. Nat. Rev. Neurol. 2013, 9, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.; Heimbach, A.; Gründemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.-L.; Roeper, J.; et al. Hereditary Parkinsonism with Dementia Is Caused by Mutations in ATP13A2, Encoding a Lysosomal Type 5 P-Type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.R.; Hadeed, A.; al-Din, A.S.N.; Wreikat, A.; Lees, A.J. Kufor Rakeb Disease: Autosomal Recessive, Levodopa-responsive Parkinsonism with Pyramidal Degeneration, Supranuclear Gaze Palsy, and Dementia. Mov. Disord. 2005, 20, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, A.; Arns, B.; Seibler, P.; Rakovic, A.; Münchau, A.; Ramirez, A.; Sue, C.M.; Klein, C. ATP13A2 Mutations Impair Mitochondrial Function in Fibroblasts from Patients with Kufor-Rakeb Syndrome. Neurobiol. Aging 2012, 33, e1–e1843. [Google Scholar] [CrossRef]
- Burke, J.E.; Dennis, E.A. Phospholipase A2 Structure/Function, Mechanism, and Signaling. J. Lipid Res. 2009, 50, S237–S242. [Google Scholar] [CrossRef]
- Kinghorn, K.J.; Castillo-Quan, J.I.; Bartolome, F.; Angelova, P.R.; Li, L.; Pope, S.; Cochemé, H.M.; Khan, S.; Asghari, S.; Bhatia, K.P.; et al. Loss of PLA2G6 Leads to Elevated Mitochondrial Lipid Peroxidation and Mitochondrial Dysfunction. Brain 2015, 138, 1801–1816. [Google Scholar] [CrossRef]
- Karkheiran, S.; Shahidi, G.A.; Walker, R.H.; Paisán-Ruiz, C. PLA2G6-Associated Dystonia-Parkinsonism: Case Report and Literature Review. Tremor Other Hyperkinetic Mov. 2015, 5, 317. [Google Scholar] [CrossRef]
- Shojaee, S.; Sina, F.; Banihosseini, S.S.; Kazemi, M.H.; Kalhor, R.; Shahidi, G.-A.; Fakhrai-Rad, H.; Ronaghi, M.; Elahi, E. Genome-Wide Linkage Analysis of a Parkinsonian-Pyramidal Syndrome Pedigree by 500 K SNP Arrays. Am. J. Hum. Genet. 2008, 82, 1375–1384. [Google Scholar] [CrossRef]
- Di Fonzo, A.; Dekker, M.C.J.; Montagna, P.; Baruzzi, A.; Yonova, E.H.; Guedes, L.C.; Szczerbinska, A.; Zhao, T.; Dubbel-Hulsman, L.O.M.; Wouters, C.H.; et al. FBXO7 Mutations Cause Autosomal Recessive, Early-Onset Parkinsonian-Pyramidal Syndrome. Neurology 2009, 72, 240–245. [Google Scholar] [CrossRef]
- Delgado-Camprubi, M.; Esteras, N.; Soutar, M.P.; Plun-Favreau, H.; Abramov, A.Y. Deficiency of Parkinson’s Disease-Related Gene Fbxo7 Is Associated with Impaired Mitochondrial Metabolism by PARP Activation. Cell Death Differ. 2017, 24, 120–131. [Google Scholar] [CrossRef]
- Conedera, S.; Apaydin, H.; Li, Y.; Yoshino, H.; Ikeda, A.; Matsushima, T.; Funayama, M.; Nishioka, K.; Hattori, N. FBXO7 Mutations in Parkinson’s Disease and Multiple System Atrophy. Neurobiol. Aging 2016, 40, e1–e192. [Google Scholar] [CrossRef] [PubMed]
- Corti, O.; Lesage, S.; Brice, A. What Genetics Tells Us About the Causes and Mechanisms of Parkinson’s Disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Darvish, H.; Bravo, P.; Tafakhori, A.; Azcona, L.J.; Ranji-Burachaloo, S.; Johari, A.H.; Paisán-Ruiz, C. Identification of a Large Homozygous VPS13C Deletion in a Patient with Early-onset Parkinsonism. Mov. Disord. 2018, 33, 1968–1970. [Google Scholar] [CrossRef]
- Matsumine, H.; Saito, M.; Shimoda-Matsubayashi, S.; Tanaka, H.; Ishikawa, A.; Nakagawa-Hattori, Y.; Yokochi, M.; Kobayashi, T.; Igarashi, S.; Takano, H.; et al. Localization of a Gene for an Autosomal Recessive Form of Juvenile Parkinsonism to Chromosome 6q25.2-27. Am. J. Hum. Genet. 1997, 60, 588–596. [Google Scholar]
- Periquet, M.; Latouche, M.; Lohmann, E.; Rawal, N.; De Michele, G.; Ricard, S.; Teive, H.; Fraix, V.; Vidailhet, M.; Nicholl, D.; et al. Parkin Mutations Are Frequent in Patients with Isolated Early-onset Parkinsonism. Brain 2003, 126, 1271–1278. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Grünewald, A.; Kasten, M.; Ziegler, A.; Klein, C. Next-Generation Phenotyping Using the Parkin Example. JAMA Neurol. 2013, 70, 1186. [Google Scholar] [CrossRef]
- Kalinderi, K.; Bostantjopoulou, S.; Fidani, L. The Genetic Background of Parkinson’s Disease: Current Progress and Future Prospects. Acta Neurol. Scand. 2016, 134, 314–326. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.K.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449.e5. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.K.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary Early-Onset Parkinson’s Disease Caused by Mutations in PINK1. Science (1979) 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The Roles of PINK1, Parkin, and Mitochondrial Fidelity in Parkinson’s Disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.-L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 Function in a Vesicular Trafficking Pathway Regulating Mitochondrial Quality Control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.-E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Jacoupy, M.; Corvol, J.-C.; Corti, O. PINK1/Parkin-Dependent Mitochondrial Surveillance: From Pleiotropy to Parkinson’s Disease. Front. Mol. Neurosci. 2017, 10, 120. [Google Scholar] [CrossRef]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin Pathway Regulates Mitochondrial Morphology. Proc. Natl. Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef]
- Arena, G.; Valente, E.M. PINK1 in the Limelight: Multiple Functions of an Eclectic Protein in Human Health and Disease. J. Pathol. 2017, 241, 251–263. [Google Scholar] [CrossRef]
- Liu, X.; Le, W. Profiling Non-Motor Symptoms in Monogenic Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 591183. [Google Scholar] [CrossRef]
- van Duijn, C.M.; Dekker, M.C.J.; Bonifati, V.; Galjaard, R.J.; Houwing-Duistermaat, J.J.; Snijders, P.J.L.M.; Testers, L.; Breedveld, G.J.; Horstink, M.; Sandkuijl, L.A.; et al. PARK7, a Novel Locus for Autosomal Recessive Early-Onset Parkinsonism, on Chromosome 1p36. Am. J. Hum. Genet. 2001, 69, 629–634. [Google Scholar] [CrossRef]
- Dolgacheva, L.P.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Abramov, A.Y. Role of DJ-1 in the Mechanism of Pathogenesis of Parkinson’s Disease. J. Bioenerg. Biomembr. 2019, 51, 175–188. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.M.; Nijland, P.G.; Drukarch, B.; de Vries, H.E.; van Horssen, J. Involvement and Interplay of Parkin, PINK1, and DJ1 in Neurodegenerative and Neuroinflammatory Disorders. Free Radic. Biol. Med. 2012, 53, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced Basal Autophagy and Impaired Mitochondrial Dynamics Due to Loss of Parkinson’s Disease-Associated Protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [PubMed]
- Piston, D.; Alvarez-Erviti, L.; Bansal, V.; Gargano, D.; Yao, Z.; Szabadkai, G.; Odell, M.; Puno, M.R.; Björkblom, B.; Maple-Grødem, J.; et al. DJ-1 Is a Redox Sensitive Adapter Protein for High Molecular Weight Complexes Involved in Regulation of Catecholamine Homeostasis. Hum. Mol. Genet. 2017, 26, 4028–4041. [Google Scholar] [CrossRef] [PubMed]
- Repici, M.; Giorgini, F. DJ-1 in Parkinson’s Disease: Clinical Insights and Therapeutic Perspectives. J. Clin. Med. 2019, 8, 1377. [Google Scholar] [CrossRef]
- Vrijsen, S.; Besora-Casals, L.; van Veen, S.; Zielich, J.; Van den Haute, C.; Hamouda, N.N.; Fischer, C.; Ghesquière, B.; Tournev, I.; Agostinis, P.; et al. ATP13A2-Mediated Endo-Lysosomal Polyamine Export Counters Mitochondrial Oxidative Stress. Proc. Natl. Acad. Sci. USA 2020, 117, 31198–31207. [Google Scholar] [CrossRef]
- Wang, R.; Tan, J.; Chen, T.; Han, H.; Tian, R.; Tan, Y.; Wu, Y.; Cui, J.; Chen, F.; Li, J.; et al. ATP13A2 Facilitates HDAC6 Recruitment to Lysosome to Promote Autophagosome–Lysosome Fusion. J. Cell Biol. 2019, 218, 267–284. [Google Scholar] [CrossRef]
- Hatori, Y.; Kanda, Y.; Nonaka, S.; Nakanishi, H.; Kitazawa, T. ATP13A2 Modifies Mitochondrial Localization of Overexpressed TOM20 to Autolysosomal Pathway. PLoS ONE 2022, 17, e0276823. [Google Scholar] [CrossRef]
- Magrinelli, F.; Mehta, S.; Di Lazzaro, G.; Latorre, A.; Edwards, M.J.; Balint, B.; Basu, P.; Kobylecki, C.; Groppa, S.; Hegde, A.; et al. Dissecting the Phenotype and Genotype of PLA2G6-Related Parkinsonism. Mov. Disord. 2022, 37, 148–161. [Google Scholar] [CrossRef]
- Morrison, K.; Witte, K.; Mayers, J.R.; Schuh, A.L.; Audhya, A. Roles of Acidic Phospholipids and Nucleotides in Regulating Membrane Binding and Activity of a Calcium-Independent Phospholipase A2 Isoform. J. Biol. Chem. 2012, 287, 38824–38834. [Google Scholar] [CrossRef]
- Beck, G.; Sugiura, Y.; Shinzawa, K.; Kato, S.; Setou, M.; Tsujimoto, Y.; Sakoda, S.; Sumi-Akamaru, H. Neuroaxonal Dystrophy in Calcium-Independent Phospholipase A2β Deficiency Results from Insufficient Remodeling and Degeneration of Mitochondrial and Presynaptic Membranes. J. Neurosci. 2011, 31, 11411–11420. [Google Scholar] [CrossRef]
- Zhou, Q.; Yen, A.; Rymarczyk, G.; Asai, H.; Trengrove, C.; Aziz, N.; Kirber, M.T.; Mostoslavsky, G.; Ikezu, T.; Wolozin, B.; et al. Impairment of PARK14-Dependent Ca2+ Signalling Is a Novel Determinant of Parkinson’s Disease. Nat. Commun. 2016, 7, 10332. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-C.; Lu, C.-S.; Weng, Y.-H.; Chen, Y.-L.; Huang, Y.-Z.; Chen, R.-S.; Cheng, Y.-C.; Huang, Y.-C.; Liu, Y.-C.; Lai, S.-C.; et al. PARK14 (D331Y) PLA2G6 Causes Early-Onset Degeneration of Substantia Nigra Dopaminergic Neurons by Inducing Mitochondrial Dysfunction, ER Stress, Mitophagy Impairment and Transcriptional Dysregulation in a Knockin Mouse Model. Mol. Neurobiol. 2019, 56, 3835–3853. [Google Scholar] [CrossRef] [PubMed]
- Sumi-Akamaru, H.; Beck, G.; Kato, S.; Mochizuki, H. Neuroaxonal Dystrophy in PLA2G6 Knockout Mice. Neuropathology 2015, 35, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Yoo, D.; Choi, J.-H.; Im, J.-H.; Kim, M.J.; Kim, H.-J.; Park, S.S.; Jeon, B. Young-Onset Parkinson’s Disease with Impulse Control Disorder Due to Novel Variants of F-Box Only Protein 7. J. Mov. Disord. 2020, 13, 225–228. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Lee, J.C.T.; Tan, E.K. Pathophysiological Mechanisms Linking F-Box Only Protein 7 (FBXO7) and Parkinson’s Disease (PD). Mutat. Res./Rev. Mutat. Res. 2018, 778, 72–78. [Google Scholar] [CrossRef]
- Schormair, B.; Kemlink, D.; Mollenhauer, B.; Fiala, O.; Machetanz, G.; Roth, J.; Berutti, R.; Strom, T.M.; Haslinger, B.; Trenkwalder, C.; et al. Diagnostic Exome Sequencing in Early-Onset Parkinson’s Disease Confirms VPS13C as a Rare Cause of Autosomal-Recessive Parkinson’s Disease. Clin. Genet. 2018, 93, 603–612. [Google Scholar] [CrossRef]
- Almeida, A.; Moncada, S.; Bolaños, J.P. Nitric Oxide Switches on Glycolysis through the AMP Protein Kinase and 6-Phosphofructo-2-Kinase Pathway. Nat. Cell Biol. 2004, 6, 45–51. [Google Scholar] [CrossRef]
- Almeida, A.; Almeida, J.; Bolaños, J.P.; Moncada, S. Different Responses of Astrocytes and Neurons to Nitric Oxide: The Role of Glycolytically Generated ATP in Astrocyte Protection. Proc. Natl. Acad. Sci. USA 2001, 98, 15294–15299. [Google Scholar] [CrossRef]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C Are Lipid Transport Proteins Differentially Localized at ER Contact Sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef]
- Go, C.D.; Knight, J.D.R.; Rajasekharan, A.; Rathod, B.; Hesketh, G.G.; Abe, K.T.; Youn, J.-Y.; Samavarchi-Tehrani, P.; Zhang, H.; Zhu, L.Y.; et al. A Proximity-Dependent Biotinylation Map of a Human Cell: An Interactive Web Resource 2019. bioRxiv 2019. [Google Scholar] [CrossRef]
- Hancock-Cerutti, W.; Wu, Z.; Xu, P.; Yadavalli, N.; Leonzino, M.; Tharkeshwar, A.K.; Ferguson, S.M.; Shadel, G.S.; De Camilli, P. ER-Lysosome Lipid Transfer Protein VPS13C/PARK23 Prevents Aberrant MtDNA-Dependent STING Signaling. J. Cell Biol. 2022, 221, e202106046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, G.; Wang, D.; Chen, H.; Su, D.; Kou, W.; Zhao, J.; Wang, X.; Wang, Z.; Ma, H.; et al. Effect of Onset Age on the Levodopa Threshold Dosage for Dyskinesia in Parkinson’s Disease. Neurol. Sci. 2022, 43, 3165–3174. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-Synuclein Gene Identified in Families with Parkinson’s Disease. Science (1979) 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Fuchs, J.; Nilsson, C.; Kachergus, J.; Munz, M.; Larsson, E.-M.; Schüle, B.; Langston, J.W.; Middleton, F.A.; Ross, O.A.; Hulihan, M.; et al. Phenotypic Variation in a Large Swedish Pedigree Due to SNCA Duplication and Triplication. Neurology 2007, 68, 916–922. [Google Scholar] [CrossRef]
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial Alpha-Synuclein Accumulation Impairs Complex I Function in Dopaminergic Neurons and Results in Increased Mitophagy In Vivo. Neurosci. Lett. 2010, 486, 235–239. [Google Scholar] [CrossRef]
- Grassi, D.; Howard, S.; Zhou, M.; Diaz-Perez, N.; Urban, N.T.; Guerrero-Given, D.; Kamasawa, N.; Volpicelli-Daley, L.A.; LoGrasso, P.; Lasmézas, C.I. Identification of a Highly Neurotoxic α-Synuclein Species Inducing Mitochondrial Damage and Mitophagy in Parkinson’s Disease. Proc. Natl. Acad. Sci. USA 2018, 115, E2634–E2643. [Google Scholar] [CrossRef]
- Puschmann, A.; Ross, O.A.; Vilariño-Güell, C.; Lincoln, S.J.; Kachergus, J.M.; Cobb, S.A.; Lindquist, S.G.; Nielsen, J.E.; Wszolek, Z.K.; Farrer, M.; et al. A Swedish Family with de Novo α-Synuclein A53T Mutation: Evidence for Early Cortical Dysfunction. Park. Relat. Disord. 2009, 15, 627–632. [Google Scholar] [CrossRef]
- Paisán-Ruíz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simón, J.; van der Brug, M.; de Munain, A.L.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the Gene Containing Mutations That Cause PARK8-Linked Parkinson’s Disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Wallings, R.; Manzoni, C.; Bandopadhyay, R. Cellular Processes Associated with LRRK2 Function and Dysfunction. FEBS J. 2015, 282, 2806–2826. [Google Scholar] [CrossRef]
- Hsieh, C.-H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St. Lawrence, E.; Schüle, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell 2016, 19, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New Insights into the Complex Role of Mitochondria in Parkinson’s Disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Mortiboys, H.; Furmston, R.; Bronstad, G.; Aasly, J.; Elliott, C.; Bandmann, O. UDCA Exerts Beneficial Effect on Mitochondrial Dysfunction in LRRK2G2019S Carriers and in Vivo. Neurology 2015, 85, 846–852. [Google Scholar] [CrossRef] [PubMed]
- Kestenbaum, M.; Alcalay, R.N. Clinical Features of LRRK2 Carriers with Parkinson’s Disease. In Advances in Neurobiology; Springer: Cham, Switzerland, 2017; pp. 31–48. [Google Scholar]
- Alcalay, R.N.; Mirelman, A.; Saunders-Pullman, R.; Tang, M.; Mejia Santana, H.; Raymond, D.; Roos, E.; Orbe-Reilly, M.; Gurevich, T.; Bar Shira, A.; et al. Parkinson Disease Phenotype in Ashkenazi Jews with and without LRRK2 G2019S Mutations. Mov. Disord. 2013, 28, 1966–1971. [Google Scholar] [CrossRef]
- Mirelman, A.; Heman, T.; Yasinovsky, K.; Thaler, A.; Gurevich, T.; Marder, K.; Bressman, S.; Bar-Shira, A.; Orr-Urtreger, A.; Giladi, N.; et al. Fall Risk and Gait in Parkinson’s Disease: The Role of the LRRK2 G2019S Mutation. Mov. Disord. 2013, 28, 1683–1690. [Google Scholar] [CrossRef]
- Trinh, J.; Guella, I.; Farrer, M.J. Disease Penetrance of Late-Onset Parkinsonism. JAMA Neurol. 2014, 71, 1535. [Google Scholar] [CrossRef]
- Ishihara, L.; Warren, L.; Gibson, R.; Amouri, R.; Lesage, S.; Dürr, A.; Tazir, M.; Wszolek, Z.K.; Uitti, R.J.; Nichols, W.C.; et al. Clinical Features of Parkinson Disease Patients with Homozygous Leucine-Rich Repeat Kinase 2 G2019S Mutations. Arch. Neurol. 2006, 63, 1250. [Google Scholar] [CrossRef]
- Strauss, K.M.; Martins, L.M.; Plun-Favreau, H.; Marx, F.P.; Kautzmann, S.; Berg, D.; Gasser, T.; Wszolek, Z.; Müller, T.; Bornemann, A.; et al. Loss of Function Mutations in the Gene Encoding Omi/HtrA2 in Parkinson’s Disease. Hum. Mol. Genet. 2005, 14, 2099–2111. [Google Scholar] [CrossRef]
- Wang, C.; Xu, Q.; Weng, L.; Zhang, Q.; Zhang, H.; Guo, J.; Tan, L.-M.; Tang, J.; Yan, X.; Tang, B. Genetic Variations of Omi/HTRA2 in Chinese Patients with Parkinson’s Disease. Brain Res. 2011, 1385, 293–297. [Google Scholar] [CrossRef]
- Casadei, N.; Sood, P.; Ulrich, T.; Fallier-Becker, P.; Kieper, N.; Helling, S.; May, C.; Glaab, E.; Chen, J.; Nuber, S.; et al. Mitochondrial Defects and Neurodegeneration in Mice Overexpressing Wild-Type or G399S Mutant HtrA2. Hum. Mol. Genet. 2016, 25, 459–471. [Google Scholar] [CrossRef]
- Vilariño-Güell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 Mutations in Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, W.; Hoppel, C.; Liu, J.; Zhu, X. Parkinson’s Disease-Associated Pathogenic VPS35 Mutation Causes Complex I Deficits. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 2791–2795. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.L.; Caldwell, M.A.; Cullen, P.J.; Liu, J.; Zhu, X. Parkinson’s Disease–Associated Mutant VPS35 Causes Mitochondrial Dysfunction by Recycling DLP1 Complexes. Nat. Med. 2016, 22, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Trinh, J.; Zeldenrust, F.M.J.; Huang, J.; Kasten, M.; Schaake, S.; Petkovic, S.; Madoev, H.; Grünewald, A.; Almuammar, S.; König, I.R.; et al. Genotype-phenotype Relations for the Parkinson’s Disease Genes SNCA, LRRK2, VPS35: MDSGene Systematic Review. Mov. Disord. 2018, 33, 1857–1870. [Google Scholar] [CrossRef]
- Vollstedt, E.; Schaake, S.; Lohmann, K.; Padmanabhan, S.; Brice, A.; Lesage, S.; Tesson, C.; Vidailhet, M.; Wurster, I.; Hentati, F.; et al. Embracing Monogenic Parkinson’s Disease: The MJFFGlobal Genetic PD Cohort. Mov. Disord. 2023, 38, 286–303. [Google Scholar] [CrossRef]
- Gao, X.-Y.; Yang, T.; Gu, Y.; Sun, X.-H. Mitochondrial Dysfunction in Parkinson’s Disease: From Mechanistic Insights to Therapy. Front. Aging Neurosci. 2022, 14, 885500. [Google Scholar] [CrossRef]
- Lee, R.G.; Sedghi, M.; Salari, M.; Shearwood, A.-M.J.; Stentenbach, M.; Kariminejad, A.; Goullee, H.; Rackham, O.; Laing, N.G.; Tajsharghi, H.; et al. Early-Onset Parkinson Disease Caused by a Mutation in CHCHD2 and Mitochondrial Dysfunction. Neurol. Genet. 2018, 4, e276. [Google Scholar] [CrossRef]
- Meng, H.; Yamashita, C.; Shiba-Fukushima, K.; Inoshita, T.; Funayama, M.; Sato, S.; Hatta, T.; Natsume, T.; Umitsu, M.; Takagi, J.; et al. Loss of Parkinson’s Disease-Associated Protein CHCHD2 Affects Mitochondrial Crista Structure and Destabilizes Cytochrome c. Nat. Commun. 2017, 8, 15500. [Google Scholar] [CrossRef]
- Puschmann, A.; Dickson, D.W.; Englund, E.; Wszolek, Z.K.; Ross, O.A. CHCHD2 and Parkinson’s Disease. Lancet Neurol. 2015, 14, 679. [Google Scholar] [CrossRef]
- Pedersen, C.C.; Lange, J.; Førland, M.G.G.; Macleod, A.D.; Alves, G.; Maple-Grødem, J. A Systematic Review of Associations between Common SNCA Variants and Clinical Heterogeneity in Parkinson’s Disease. NPJ Park. Dis. 2021, 7, 54. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K. α-Synuclein and Mitochondria: Partners in Crime? Neurotherapeutics 2013, 10, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Vicario, M.; Cieri, D.; Brini, M.; Calì, T. The Close Encounter Between Alpha-Synuclein and Mitochondria. Front. Neurosci. 2018, 12, 388. [Google Scholar] [CrossRef] [PubMed]
- Thorne, N.J.; Tumbarello, D.A. The Relationship of Alpha-Synuclein to Mitochondrial Dynamics and Quality Control. Front. Mol. Neurosci. 2022, 15, 947191. [Google Scholar] [CrossRef]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. α-Synuclein Binds to TOM20 and Inhibits Mitochondrial Protein Import in Parkinson’s Disease. Sci. Transl. Med. 2016, 8, 342ra78. [Google Scholar] [CrossRef]
- Hunn, B.H.M.; Cragg, S.J.; Bolam, J.P.; Spillantini, M.-G.; Wade-Martins, R. Impaired Intracellular Trafficking Defines Early Parkinson’s Disease. Trends Neurosci. 2015, 38, 178–188. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein Locus Triplication Causes Parkinson’s Disease. Science (1979) 2003, 302, 841. [Google Scholar] [CrossRef]
- Martin, I.; Kim, J.W.; Dawson, V.L.; Dawson, T.M. LRRK2 Pathobiology in Parkinson’s Disease. J. Neurochem. 2014, 131, 554–565. [Google Scholar] [CrossRef]
- Kang, U.; Marto, J.A. Leucine-rich Repeat Kinase 2 and Parkinson’s Disease. Proteomics 2017, 17, 1600092. [Google Scholar] [CrossRef]
- Angeles, D.C.; Gan, B.-H.; Onstead, L.; Zhao, Y.; Lim, K.-L.; Dachsel, J.; Melrose, H.; Farrer, M.; Wszolek, Z.K.; Dickson, D.W.; et al. Mutations in LRRK2 Increase Phosphorylation of Peroxiredoxin 3 Exacerbating Oxidative Stress-Induced Neuronal Death. Hum. Mutat. 2011, 32, 1390–1397. [Google Scholar] [CrossRef]
- Biskup, S.; Moore, D.J.; Celsi, F.; Higashi, S.; West, A.B.; Andrabi, S.A.; Kurkinen, K.; Yu, S.; Savitt, J.M.; Waldvogel, H.J.; et al. Localization of LRRK2 to Membranous and Vesicular Structures in Mammalian Brain. Ann. Neurol. 2006, 60, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Yue, M.; Hinkle, K.M.; Davies, P.; Trushina, E.; Fiesel, F.C.; Christenson, T.A.; Schroeder, A.S.; Zhang, L.; Bowles, E.; Behrouz, B.; et al. Progressive Dopaminergic Alterations and Mitochondrial Abnormalities in LRRK2 G2019S Knock-in Mice. Neurobiol. Dis. 2015, 78, 172–195. [Google Scholar] [CrossRef] [PubMed]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics Reveals That Parkinson’s Disease Kinase LRRK2 Regulates a Subset of Rab GTPases. eLife 2016, 5, e12813. [Google Scholar] [CrossRef]
- Dagda, R.K.; Chu, C.T. Mitochondrial Quality Control: Insights on How Parkinson’s Disease Related Genes PINK1, Parkin, and Omi/HtrA2 Interact to Maintain Mitochondrial Homeostasis. J. Bioenerg. Biomembr. 2009, 41, 473–479. [Google Scholar] [CrossRef]
- Verhagen, A.M.; Silke, J.; Ekert, P.G.; Pakusch, M.; Kaufmann, H.; Connolly, L.M.; Day, C.L.; Tikoo, A.; Burke, R.; Wrobel, C.; et al. HtrA2 Promotes Cell Death through Its Serine Protease Activity and Its Ability to Antagonize Inhibitor of Apoptosis Proteins. J. Biol. Chem. 2002, 277, 445–454. [Google Scholar] [CrossRef]
- Patil, K.S.; Basak, I.; Lee, S.; Abdullah, R.; Larsen, J.P.; Møller, S.G. PARK13 Regulates PINK1 and Subcellular Relocation Patterns under Oxidative Stress in Neurons. J. Neurosci. Res. 2014, 92, 1167–1177. [Google Scholar] [CrossRef]
- Fitzgerald, J.C.; Camprubi, M.D.; Dunn, L.; Wu, H.-C.; Ip, N.Y.; Kruger, R.; Martins, L.M.; Wood, N.W.; Plun-Favreau, H. Phosphorylation of HtrA2 by Cyclin-Dependent Kinase-5 Is Important for Mitochondrial Function. Cell Death Differ. 2012, 19, 257–266. [Google Scholar] [CrossRef]
- Sharma, M.; Ioannidis, J.P.A.; Aasly, J.O.; Annesi, G.; Brice, A.; Bertram, L.; Bozi, M.; Barcikowska, M.; Crosiers, D.; Clarke, C.E.; et al. A Multi-Centre Clinico-Genetic Analysis of the VPS35 Gene in Parkinson Disease Indicates Reduced Penetrance for Disease-Associated Variants. J. Med. Genet. 2012, 49, 721–726. [Google Scholar] [CrossRef]
- Wider, C.; Skipper, L.; Solida, A.; Brown, L.; Farrer, M.; Dickson, D.; Wszolek, Z.K.; Vingerhoets, F.J.G. Autosomal Dominant Dopa-Responsive Parkinsonism in a Multigenerational Swiss Family. Park. Relat. Disord. 2008, 14, 465–470. [Google Scholar] [CrossRef]
- Tang, F.-L.; Liu, W.; Hu, J.-X.; Erion, J.R.; Ye, J.; Mei, L.; Xiong, W.-C. VPS35 Deficiency or Mutation Causes Dopaminergic Neuronal Loss by Impairing Mitochondrial Fusion and Function. Cell Rep. 2015, 12, 1631–1643. [Google Scholar] [CrossRef]
- Paravicini, G.; Horazdovsky, B.F.; Emr, S.D. Alternative Pathways for the Sorting of Soluble Vacuolar Proteins in Yeast: A Vps35 Null Mutant Missorts and Secretes Only a Subset of Vacuolar Hydrolases. Mol. Biol. Cell 1992, 3, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Haft, C.R.; Sierra, M.d.l.L.; Bafford, R.; Lesniak, M.A.; Barr, V.A.; Taylor, S.I. Human Orthologs of Yeast Vacuolar Protein Sorting Proteins Vps26, 29, and 35: Assembly into Multimeric Complexes. Mol. Biol. Cell 2000, 11, 4105–4116. [Google Scholar] [CrossRef] [PubMed]
- Funayama, M.; Ohe, K.; Amo, T.; Furuya, N.; Yamaguchi, J.; Saiki, S.; Li, Y.; Ogaki, K.; Ando, M.; Yoshino, H.; et al. CHCHD2 Mutations in Autosomal Dominant Late-Onset Parkinson’s Disease: A Genome-Wide Linkage and Sequencing Study. Lancet Neurol. 2015, 14, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Li, K. CHCHD2 and Parkinson’s Disease. Lancet Neurol. 2015, 14, 679–680. [Google Scholar] [CrossRef]
- Niemann, N.; Jankovic, J. Juvenile Parkinsonism: Differential Diagnosis, Genetics, and Treatment. Park. Relat. Disord. 2019, 67, 74–89. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I.; et al. CAG Expansions in a Novel Gene for Machado-Joseph Disease at Chromosome 14q32.1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef]
- Mendonça, N.; França, M.C.; Gonçalves, A.F.; Januário, C. Clinical Features of Machado-Joseph Disease. Adv. Exp. Med. Biol. 2018, 1049, 255–273. [Google Scholar]
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar Ataxia. Nat. Rev. Dis. Primers 2019, 5, 24. [Google Scholar] [CrossRef]
- Harmuth, T.; Weber, J.J.; Zimmer, A.J.; Sowa, A.S.; Schmidt, J.; Fitzgerald, J.C.; Schöls, L.; Riess, O.; Hübener-Schmid, J. Mitochondrial Dysfunction in Spinocerebellar Ataxia Type 3 Is Linked to VDAC1 Deubiquitination. Int. J. Mol. Sci. 2022, 23, 5933. [Google Scholar] [CrossRef]
- Fossale, E.; Wolf, P.; Espinola, J.A.; Lubicz-Nawrocka, T.; Teed, A.M.; Gao, H.; Rigamonti, D.; Cattaneo, E.; MacDonald, M.E.; Cotman, S.L. Membrane Trafficking and Mitochondrial Abnormalities Precede Subunit c Deposition in a Cerebellar Cell Model of Juvenile Neuronal Ceroid Lipofuscinosis. BMC Neurosci. 2004, 5, 57. [Google Scholar] [CrossRef]
- Mirza, M.; Vainshtein, A.; DiRonza, A.; Chandrachud, U.; Haslett, L.J.; Palmieri, M.; Storch, S.; Groh, J.; Dobzinski, N.; Napolitano, G.; et al. The CLN3 Gene and Protein: What We Know. Mol. Genet. Genom. Med. 2019, 7, e859. [Google Scholar] [CrossRef] [PubMed]
- Rha, A.K.; Maguire, A.S.; Martin, D.R. GM1 Gangliosidosis: Mechanisms and Management. Appl. Clin. Genet. 2021, 14, 209–233. [Google Scholar] [CrossRef] [PubMed]
- Davidzon, G.; Greene, P.; Mancuso, M.; Klos, K.J.; Ahlskog, J.E.; Hirano, M.; DiMauro, S. Early-onset Familial Parkinsonism Due to POLG Mutations. Ann. Neurol. 2006, 59, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.H.; Ng, Y.S.; Gorman, G.S.; Alston, C.L.; Blakely, E.L.; Schaefer, A.M.; Chinnery, P.F.; Burn, D.J.; Taylor, R.W.; McFarland, R.; et al. Clinical, Genetic, and Radiological Features of Extrapyramidal Movement Disorders in Mitochondrial Disease. JAMA Neurol. 2016, 73, 668. [Google Scholar] [CrossRef]
- Rahman, S.; Copeland, W.C. POLG-Related Disorders and Their Neurological Manifestations. Nat. Rev. Neurol. 2019, 15, 40–52. [Google Scholar] [CrossRef]
- Copeland, W.C.; Longley, M.J. DNA Polymerase Gamma in Mitochondrial DNA Replication and Repair. Sci. World J. 2003, 3, 34–44. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. Mutant Ataxin-3 Promotes the Autophagic Degradation of Parkin. Autophagy 2011, 7, 233–234. [Google Scholar] [CrossRef]
- Durcan, T.M.; Kontogiannea, M.; Thorarinsdottir, T.; Fallon, L.; Williams, A.J.; Djarmati, A.; Fantaneanu, T.; Paulson, H.L.; Fon, E.A. The Machado–Joseph Disease-Associated Mutant Form of Ataxin-3 Regulates Parkin Ubiquitination and Stability. Hum. Mol. Genet. 2011, 20, 141–154. [Google Scholar] [CrossRef]
- Durcan, T.M.; Kontogiannea, M.; Bedard, N.; Wing, S.S.; Fon, E.A. Ataxin-3 Deubiquitination Is Coupled to Parkin Ubiquitination via E2 Ubiquitin-Conjugating Enzyme. J. Biol. Chem. 2012, 287, 531–541. [Google Scholar] [CrossRef]
- Paulson, H.; Shakkottai, V. Spinocerebellar Ataxia Type 3. In GeneReviews®; 10 October 1998 [updated 4 June 2020]; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993–2025. [Google Scholar]
- Kuo, M.; Tai, C.; Tseng, S.; Wu, R. Long-term Efficacy of Bilateral Subthalamic Deep Brain Stimulation in the Parkinsonism of SCA 3: A Rare Case Report. Eur. J. Neurol. 2022, 29, 2544–2547. [Google Scholar] [CrossRef]
- Palmer, D.N.; Barry, L.A.; Tyynelä, J.; Cooper, J.D. NCL Disease Mechanisms. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Ostergaard, J.R.; Nelvagal, H.R.; Cooper, J.D. Top-down and Bottom-up Propagation of Disease in the Neuronal Ceroid Lipofuscinoses. Front. Neurol. 2022, 13, 1061363. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.J. Recent Insights into the Networking of CLN Genes and Proteins in Mammalian Cells. J. Neurochem. 2023, 165, 643–659. [Google Scholar] [CrossRef]
- Dozières-Puyravel, B.; Nasser, H.; Elmaleh-Bergès, M.; Lopez Hernandez, E.; Gelot, A.; Ilea, A.; Delanoë, C.; Puech, J.; Caillaud, C.; Pichard, S.; et al. Paediatric-onset Neuronal Ceroid Lipofuscinosis: First Symptoms and Presentation at Diagnosis. Dev. Med. Child. Neurol. 2020, 62, 528–530. [Google Scholar] [CrossRef]
- Relton, E.L.; Roth, N.J.; Yasa, S.; Kaleem, A.; Hermey, G.; Minnis, C.J.; Mole, S.E.; Shelkovnikova, T.; Lefrancois, S.; McCormick, P.J.; et al. The Batten Disease Protein CLN3 Is Important for Stress Granules Dynamics and Translational Activity. J. Biol. Chem. 2023, 299, 104649. [Google Scholar] [CrossRef]
- Luiro, K.; Kopra, O.; Blom, T.; Gentile, M.; Mitchison, H.M.; Hovatta, I.; Törnquist, K.; Jalanko, A. Batten Disease (JNCL) Is Linked to Disturbances in Mitochondrial, Cytoskeletal, and Synaptic Compartments. J. Neurosci. Res. 2006, 84, 1124–1138. [Google Scholar] [CrossRef]
- Metcalf, D.J.; Calvi, A.A.; Seaman, M.N.; Mitchison, H.M.; Cutler, D.F. Loss of the Batten Disease Gene CLN3 Prevents Exit from the TGN of the Mannose 6-Phosphate Receptor. Traffic 2008, 9, 1905–1914. [Google Scholar] [CrossRef]
- Specchio, N.; Ferretti, A.; Trivisano, M.; Pietrafusa, N.; Pepi, C.; Calabrese, C.; Livadiotti, S.; Simonetti, A.; Rossi, P.; Curatolo, P.; et al. Neuronal Ceroid Lipofuscinosis: Potential for Targeted Therapy. Drugs 2021, 81, 101–123. [Google Scholar] [CrossRef]
- Bartsch, U.; Storch, S. Experimental Therapeutic Approaches for the Treatment of Retinal Pathology in Neuronal Ceroid Lipofuscinoses. Front. Neurol. 2022, 13, 866983. [Google Scholar] [CrossRef]
- Takamura, A.; Higaki, K.; Kajimaki, K.; Otsuka, S.; Ninomiya, H.; Matsuda, J.; Ohno, K.; Suzuki, Y.; Nanba, E. Enhanced Autophagy and Mitochondrial Aberrations in Murine GM1-Gangliosidosis. Biochem. Biophys. Res. Commun. 2008, 367, 616–622. [Google Scholar] [CrossRef]
- Blumenreich, S.; Barav, O.B.; Jenkins, B.J.; Futerman, A.H. Lysosomal Storage Disorders Shed Light on Lysosomal Dysfunction in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 4966. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Singh, G.; Williams, C.K.; Singh, V. GM1 Ganglioside Modifies Microglial and Neuroinflammatory Responses to α-Synuclein in the Rat AAV-A53T α-Synuclein Model of Parkinson’s Disease. Mol. Cell. Neurosci. 2022, 120, 103729. [Google Scholar] [CrossRef] [PubMed]
- Nicoli, E.-R.; Annunziata, I.; d’Azzo, A.; Platt, F.M.; Tifft, C.J.; Stepien, K.M. GM1 Gangliosidosis—A Mini-Review. Front. Genet. 2021, 12, 734878. [Google Scholar] [CrossRef] [PubMed]
- Van Goethem, G.; Dermaut, B.; Löfgren, A.; Martin, J.-J.; Van Broeckhoven, C. Mutation of POLG Is Associated with Progressive External Ophthalmoplegia Characterized by MtDNA Deletions. Nat. Genet. 2001, 28, 211–212. [Google Scholar] [CrossRef]
- Luoma, P.; Melberg, A.; Rinne, J.O.; Kaukonen, J.A.; Nupponen, N.N.; Chalmers, R.M.; Oldfors, A.; Rautakorpi, I.; Peltonen, L.; Majamaa, K.; et al. Parkinsonism, Premature Menopause, and Mitochondrial DNA Polymerase γ Mutations: Clinical and Molecular Genetic Study. Lancet 2004, 364, 875–882. [Google Scholar] [CrossRef]
- Luoma, P.T.; Eerola, J.; Ahola, S.; Hakonen, A.H.; Hellström, O.; Kivistö, K.T.; Tienari, P.J.; Suomalainen, A. Mitochondrial DNA Polymerase Gamma Variants in Idiopathic Sporadic Parkinson Disease. Neurology 2007, 69, 1152–1159. [Google Scholar] [CrossRef]
- Eerola, J.; Luoma, P.T.; Peuralinna, T.; Scholz, S.; Paisan-Ruiz, C.; Suomalainen, A.; Singleton, A.B.; Tienari, P.J. POLG1 Polyglutamine Tract Variants Associated with Parkinson’s Disease. Neurosci. Lett. 2010, 477, 1–5. [Google Scholar] [CrossRef]
- Chan, S.S.L.; Copeland, W.C. DNA Polymerase Gamma and Mitochondrial Disease: Understanding the Consequence of POLG Mutations. Biochim. Biophys. Acta (BBA)-Bioenerg. 2009, 1787, 312–319. [Google Scholar] [CrossRef]
- Longley, M.J.; Prasad, R.; Srivastava, D.K.; Wilson, S.H.; Copeland, W.C. Identification of 5′-Deoxyribose Phosphate Lyase Activity in Human DNA Polymerase γ and Its Role in Mitochondrial Base Excision Repair In Vitro. Proc. Natl. Acad. Sci. USA 1998, 95, 12244–12248. [Google Scholar] [CrossRef]
- Miguel, R.; Gago, M.F.; Martins, J.; Barros, P.; Vale, J.; Rosas, M.J. POLG1-Related Levodopa-Responsive Parkinsonism. Clin. Neurol. Neurosurg. 2014, 126, 47–54. [Google Scholar] [CrossRef]
- Peter, B.; Falkenberg, M. TWINKLE and Other Human Mitochondrial DNA Helicases: Structure, Function and Disease. Genes 2020, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Spelbrink, J.N.; Li, F.-Y.; Tiranti, V.; Nikali, K.; Yuan, Q.-P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human Mitochondrial DNA Deletions Associated with Mutations in the Gene Encoding Twinkle, a Phage T7 Gene 4-like Protein Localized in Mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.P.; Munoz, D.G.; Lang, A.E. Twinkle-Associated Familial Parkinsonism with Lewy Pathology. Neurology 2020, 95, 644–647. [Google Scholar] [CrossRef]
- Hedera, P. Hereditary Spastic Paraplegia Overview. In GeneReviews®; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Atorino, L.; Silvestri, L.; Koppen, M.; Cassina, L.; Ballabio, A.; Marconi, R.; Langer, T.; Casari, G. Loss of M-AAA Protease in Mitochondria Causes Complex I Deficiency and Increased Sensitivity to Oxidative Stress in Hereditary Spastic Paraplegia. J. Cell Biol. 2003, 163, 777–787. [Google Scholar] [CrossRef] [PubMed]
- Koppen, M.; Metodiev, M.D.; Casari, G.; Rugarli, E.I.; Langer, T. Variable and Tissue-Specific Subunit Composition of Mitochondrial m-AAA Protease Complexes Linked to Hereditary Spastic Paraplegia. Mol. Cell Biol. 2007, 27, 758–767. [Google Scholar] [CrossRef]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.-C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 Processing and Mitochondrial Fusion by m-AAA Protease Isoenzymes and OMA1. J. Cell Biol. 2009, 187, 1023–1036. [Google Scholar] [CrossRef]
- Patron, M.; Sprenger, H.-G.; Langer, T. M-AAA Proteases, Mitochondrial Calcium Homeostasis and Neurodegeneration. Cell Res. 2018, 28, 296–306. [Google Scholar] [CrossRef]
- Shanmughapriya, S.; Rajan, S.; Hoffman, N.E.; Higgins, A.M.; Tomar, D.; Nemani, N.; Hines, K.J.; Smith, D.J.; Eguchi, A.; Vallem, S.; et al. SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Mol. Cell 2015, 60, 47–62. [Google Scholar] [CrossRef]
- Bernardi, P.; Forte, M. Commentary: SPG7 Is an Essential and Conserved Component of the Mitochondrial Permeability Transition Pore. Front. Physiol. 2015, 6, 320. [Google Scholar] [CrossRef]
- Maltecca, F.; Aghaie, A.; Schroeder, D.G.; Cassina, L.; Taylor, B.A.; Phillips, S.J.; Malaguti, M.; Previtali, S.; Guénet, J.-L.; Quattrini, A.; et al. The Mitochondrial Protease AFG3L2 Is Essential for Axonal Development. J. Neurosci. 2008, 28, 2827–2836. [Google Scholar] [CrossRef]
- Wedding, I.M.; Koht, J.; Tran, G.T.; Misceo, D.; Selmer, K.K.; Holmgren, A.; Frengen, E.; Bindoff, L.; Tallaksen, C.M.E.; Tzoulis, C. Spastic Paraplegia Type 7 Is Associated with Multiple Mitochondrial DNA Deletions. PLoS ONE 2014, 9, e86340. [Google Scholar] [CrossRef] [PubMed]
- Coarelli, G.; Schule, R.; van de Warrenburg, B.P.C.; De Jonghe, P.; Ewenczyk, C.; Martinuzzi, A.; Synofzik, M.; Hamer, E.G.; Baets, J.; Anheim, M.; et al. Loss of Paraplegin Drives Spasticity Rather than Ataxia in a Cohort of 241 Patients with SPG7. Neurology 2019, 92, e2679–e2690. [Google Scholar] [CrossRef]
- Pedroso, J.L.; Vale, T.C.; Bueno, F.L.; Marussi, V.H.R.; do Amaral, L.L.F.; França, M.C.; Barsottini, O.G. SPG7 with Parkinsonism Responsive to Levodopa and Dopaminergic Deficit. Park. Relat. Disord. 2018, 47, 88–90. [Google Scholar] [CrossRef] [PubMed]
- Magri, S.; Fracasso, V.; Plumari, M.; Alfei, E.; Ghezzi, D.; Gellera, C.; Rusmini, P.; Poletti, A.; Di Bella, D.; Elia, A.E.; et al. Concurrent AFG3L2 and SPG7 Mutations Associated with Syndromic Parkinsonism and Optic Atrophy with Aberrant OPA1 Processing and Mitochondrial Network Fragmentation. Hum. Mutat. 2018, 39, 2060–2071. [Google Scholar] [CrossRef] [PubMed]
- De la Casa-Fages, B.; Fernández-Eulate, G.; Gamez, J.; Barahona-Hernando, R.; Morís, G.; García-Barcina, M.; Infante, J.; Zulaica, M.; Fernández-Pelayo, U.; Muñoz-Oreja, M.; et al. Parkinsonism and Spastic Paraplegia Type 7: Expanding the Spectrum of Mitochondrial Parkinsonism. Mov. Disord. 2019, 34, 1547–1561. [Google Scholar] [CrossRef]
- Trinh, J.; Lohmann, K.; Baumann, H.; Balck, A.; Borsche, M.; Brüggemann, N.; Dure, L.; Dean, M.; Volkmann, J.; Tunc, S.; et al. Utility and Implications of Exome Sequencing in Early-onset Parkinson’s Disease. Mov. Disord. 2019, 34, 133–137. [Google Scholar] [CrossRef]
- Vaglini, F.; Viaggi, C.; Piro, V.; Pardini, C.; Gerace, C.; Scarselli, M.; Corsini, G.U. Acetaldehyde and Parkinsonism: Role of CYP450 2E1. Front. Behav. Neurosci. 2013, 7, 50607. [Google Scholar] [CrossRef]
- Fasciani, I.; Petragnano, F.; Aloisi, G.; Marampon, F.; Rossi, M.; Coppolino, M.F.; Rossi, R.; Longoni, B.; Scarselli, M.; Maggio, R. A New Threat to Dopamine Neurons: The Downside of Artificial Light. Neuroscience 2020, 432, 216–228. [Google Scholar] [CrossRef]
- Nandipati, S.; Litvan, I. Environmental Exposures and Parkinson’s Disease. Int. J. Environ. Res. Public Health 2016, 13, 881. [Google Scholar] [CrossRef]
- Rossi, M.; Scarselli, M.; Fasciani, I.; Maggio, R.; Giorgi, F. Dichlorodiphenyltrichloroethane (DDT) Induced Extracellular Vesicle Formation: A Potential Role in Organochlorine Increased Risk of Parkinson’s Disease. Acta Neurobiol. Exp. (Wars) 2017, 77, 113–117. [Google Scholar] [CrossRef]
- Wan, N.; Lin, G. Parkinson’s Disease and Pesticides Exposure: New Findings from a Comprehensive Study in Nebraska, USA. J. Rural. Health 2016, 32, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in Humans Due to a Product of Meperidine-Analog Synthesis. Science (1979) 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Schildknecht, S.; Di Monte, D.A.; Pape, R.; Tieu, K.; Leist, M. Tipping Points and Endogenous Determinants of Nigrostriatal Degeneration by MPTP. Trends Pharmacol. Sci. 2017, 38, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Jackson-Lewis, V.; Blesa, J.; Przedborski, S. Animal Models of Parkinson’s Disease. Park. Relat. Disord. 2012, 18, S183–S185. [Google Scholar] [CrossRef]
- Fabre, E.; Monserrat, J.; Herrero, A.; Barja, G.; Leret, M.L. Effect of MPTP on Brain Mitochondrial H2O2 and ATP Production and on Dopamine and DOPAC in the Striatum. J. Physiol. Biochem. 1999, 55, 325–331. [Google Scholar] [CrossRef]
- Schildknecht, S.; Pape, R.; Meiser, J.; Karreman, C.; Strittmatter, T.; Odermatt, M.; Cirri, E.; Friemel, A.; Ringwald, M.; Pasquarelli, N.; et al. Preferential Extracellular Generation of the Active Parkinsonian Toxin MPP + by Transporter-Independent Export of the Intermediate MPDP+. Antioxid. Redox Signal 2015, 23, 1001–1016. [Google Scholar] [CrossRef]
- Cannon, J.R.; Tapias, V.; Na, H.M.; Honick, A.S.; Drolet, R.E.; Greenamyre, J.T. A Highly Reproducible Rotenone Model of Parkinson’s Disease. Neurobiol. Dis. 2009, 34, 279–290. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, H.; Guo, X.; Ge, D.; Shi, Y.; Lu, X.; Lu, J.; Chen, J.; Ding, F.; Zhang, Q. Involvement of Akt/MTOR in the Neurotoxicity of Rotenone-Induced Parkinson’s Disease Models. Int. J. Environ. Res. Public Health 2019, 16, 3811. [Google Scholar] [CrossRef]
- Emmrich, J.V.; Hornik, T.C.; Neher, J.J.; Brown, G.C. Rotenone Induces Neuronal Death by Microglial Phagocytosis of Neurons. FEBS J. 2013, 280, 5030–5038. [Google Scholar] [CrossRef]
- Carli, M.; Vaglini, F.; Risaliti, E.; Citi, G.; Masini, M.; Kolachalam, S.; Maggio, R.; Corsini, G.U.; Novelli, M.; De Tata, V.; et al. β-Cells Different Vulnerability to the Parkinsonian Neurotoxins Rotenone, 1-Methyl-4-Phenylpyridinium (MPP+) and 6-Hydroxydopamine (6-OHDA). Pharmaceuticals 2021, 14, 767. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Chen, M.; Du, T.; Duan, C.; Gao, G.; Yang, H. The Novel Mechanism of Rotenone-Induced α-Synuclein Phosphorylation via Reduced Protein Phosphatase 2A Activity. Int. J. Biochem. Cell Biol. 2016, 75, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Batandier, C.; Guigas, B.; Detaille, D.; El-Mir, M.; Fontaine, E.; Rigoulet, M.; Leverve, X.M. The ROS Production Induced by a Reverse-Electron Flux at Respiratory-Chain Complex 1 Is Hampered by Metformin. J. Bioenerg. Biomembr. 2006, 38, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Su, C.; Qiao, C.; Bian, Y.; Ding, J.; Hu, G. Metformin Prevents Dopaminergic Neuron Death in MPTP/P-Induced Mouse Model of Parkinson’s Disease via Autophagy and Mitochondrial ROS Clearance. Int. J. Neuropsychopharmacol. 2016, 19, pyw047. [Google Scholar] [CrossRef] [PubMed]
- Greenamyre, J.T.; Hastings, T.G. Parkinson’s--Divergent Causes, Convergent Mechanisms. Science (1979) 2004, 304, 1120–1122. [Google Scholar] [CrossRef]
- Trist, B.G.; Hare, D.J.; Double, K.L. Oxidative Stress in the Aging Substantia Nigra and the Etiology of Parkinson’s Disease. Aging Cell 2019, 18, e13031. [Google Scholar] [CrossRef]
- Arriagada, C.; Paris, I.; Sanchez de las Matas, M.J.; Martinez-Alvarado, P.; Cardenas, S.; Castañeda, P.; Graumann, R.; Perez-Pastene, C.; Olea-Azar, C.; Couve, E.; et al. On the Neurotoxicity Mechanism of Leukoaminochrome O-Semiquinone Radical Derived from Dopamine Oxidation: Mitochondria Damage, Necrosis, and Hydroxyl Radical Formation. Neurobiol. Dis. 2004, 16, 468–477. [Google Scholar] [CrossRef]
- Schildknecht, S.; Gerding, H.R.; Karreman, C.; Drescher, M.; Lashuel, H.A.; Outeiro, T.F.; Di Monte, D.A.; Leist, M. Oxidative and Nitrative Alpha-synuclein Modifications and Proteostatic Stress: Implications for Disease Mechanisms and Interventions in Synucleinopathies. J. Neurochem. 2013, 125, 491–511. [Google Scholar] [CrossRef]
- Muñoz, Y.; Carrasco, C.M.; Campos, J.D.; Aguirre, P.; Núñez, M.T. Parkinson’s Disease: The Mitochondria-Iron Link. Park. Dis. 2016, 2016, 049108. [Google Scholar] [CrossRef]
- Sanders, L.H.; McCoy, J.; Hu, X.; Mastroberardino, P.G.; Dickinson, B.C.; Chang, C.J.; Chu, C.T.; Van Houten, B.; Greenamyre, J.T. Mitochondrial DNA Damage: Molecular Marker of Vulnerable Nigral Neurons in Parkinson’s Disease. Neurobiol. Dis. 2014, 70, 214–223. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I.; Ogawa, N. Dopamine- or L-DOPA-Induced Neurotoxicity: The Role of Dopamine Quinone Formation and Tyrosinase in a Model of Parkinson’s Disease. Neurotox. Res. 2003, 5, 165–176. [Google Scholar] [CrossRef]
- Simola, N.; Morelli, M.; Carta, A.R. The 6-Hydroxydopamine Model of Parkinson’s Disease. Neurotox. Res. 2007, 11, 151–167. [Google Scholar] [CrossRef]
- Lehmensiek, V.; Tan, E.-M.; Liebau, S.; Lenk, T.; Zettlmeisl, H.; Schwarz, J.; Storch, A. Dopamine Transporter-Mediated Cytotoxicity of 6-Hydroxydopamine in Vitro Depends on Expression of Mutant α-Synucleins Related to Parkinson’s Disease. Neurochem. Int. 2006, 48, 329–340. [Google Scholar] [CrossRef]
- Blesa, J.; Przedborski, S. Parkinson’s Disease: Animal Models and Dopaminergic Cell Vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Perier, C.; Bové, J.; Vila, M.; Przedborski, S. The Rotenone Model of Parkinson’s Disease. Trends Neurosci. 2003, 26, 345–346. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.-K.; Cheng, C.-H.; Chen, S.-D.; Liou, C.-W.; Huang, C.-R.; Chuang, Y.-C. Mitochondrial Dysfunction and Oxidative Stress Promote Apoptotic Cell Death in the Striatum via Cytochrome c/Caspase-3 Signaling Cascade Following Chronic Rotenone Intoxication in Rats. Int. J. Mol. Sci. 2012, 13, 8722–8739. [Google Scholar] [CrossRef] [PubMed]
- Ransom, B.R.; Kunis, D.M.; Irwin, I.; Langston, J.W. Astrocytes Convert the Parkinsonism Inducing Neurotoxin, MPTP, to Its Active Metabolite, MPP+. Neurosci. Lett. 1987, 75, 323–328. [Google Scholar] [CrossRef]
- Blum, D.; Torch, S.; Lambeng, N.; Nissou, M.-F.; Benabid, A.-L.; Sadoul, R.; Verna, J.-M. Molecular Pathways Involved in the Neurotoxicity of 6-OHDA, Dopamine and MPTP: Contribution to the Apoptotic Theory in Parkinson’s Disease. Prog. Neurobiol. 2001, 65, 135–172. [Google Scholar] [CrossRef]
- Yan, A.; Liu, Z.; Song, L.; Wang, X.; Zhang, Y.; Wu, N.; Lin, J.; Liu, Y.; Liu, Z. Idebenone Alleviates Neuroinflammation and Modulates Microglial Polarization in LPS-Stimulated BV2 Cells and MPTP-Induced Parkinson’s Disease Mice. Front. Cell Neurosci. 2019, 12, 529. [Google Scholar] [CrossRef]
- Beal, M.F.; Matthews, R.T.; Tieleman, A.; Shults, C.W. Coenzyme Q10 Attenuates the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine (MPTP) Induced Loss of Striatal Dopamine and Dopaminergic Axons in Aged Mice. Brain Res. 1998, 783, 109–114. [Google Scholar] [CrossRef]
- Trinh, D.; Israwi, A.R.; Arathoon, L.R.; Gleave, J.A.; Nash, J.E. The Multi-faceted Role of Mitochondria in the Pathology of Parkinson’s Disease. J. Neurochem. 2021, 156, 715–752. [Google Scholar] [CrossRef]
- Biju, K.C.; Evans, R.C.; Shrestha, K.; Carlisle, D.C.B.; Gelfond, J.; Clark, R.A. Methylene Blue Ameliorates Olfactory Dysfunction and Motor Deficits in a Chronic MPTP/Probenecid Mouse Model of Parkinson’s Disease. Neuroscience 2018, 380, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Koch, W.; Elstner, M.; Schombacher, Y.; Bender, J.; Moeschl, M.; Gekeler, F.; Müller-Myhsok, B.; Gasser, T.; Tatsch, K.; et al. Creatine Supplementation in Parkinson Disease: A Placebo-Controlled Randomized Pilot Trial. Neurology 2006, 67, 1262–1264. [Google Scholar] [CrossRef] [PubMed]
- Andres, R.H.; Huber, A.W.; Schlattner, U.; Pérez-Bouza, A.; Krebs, S.H.; Seiler, R.W.; Wallimann, T.; Widmer, H.R. Effects of Creatine Treatment on the Survival of Dopaminergic Neurons in Cultured Fetal Ventral Mesencephalic Tissue. Neuroscience 2005, 133, 701–713. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, D.; Jiang, P.; Tang, X.; Lang, Q.; Yu, Q.; Zhang, S.; Che, Y.; Feng, X. Resveratrol Synergizes with Low Doses of L-DOPA to Improve MPTP-Induced Parkinson Disease in Mice. Behav. Brain Res. 2019, 367, 10–18. [Google Scholar] [CrossRef]
- Palle, S.; Neerati, P. Improved Neuroprotective Effect of Resveratrol Nanoparticles as Evinced by Abrogation of Rotenone-Induced Behavioral Deficits and Oxidative and Mitochondrial Dysfunctions in Rat Model of Parkinson’s Disease. Naunyn Schmiedebergs Arch. Pharmacol. 2018, 391, 445–453. [Google Scholar] [CrossRef]
- Abdelkader, N.F.; Safar, M.M.; Salem, H.A. Ursodeoxycholic Acid Ameliorates Apoptotic Cascade in the Rotenone Model of Parkinson’s Disease: Modulation of Mitochondrial Perturbations. Mol. Neurobiol. 2016, 53, 810–817. [Google Scholar] [CrossRef]
- Rosa, A.I.; Fonseca, I.; Nunes, M.J.; Moreira, S.; Rodrigues, E.; Carvalho, A.N.; Rodrigues, C.M.P.; Gama, M.J.; Castro-Caldas, M. Novel Insights into the Antioxidant Role of Tauroursodeoxycholic Acid in Experimental Models of Parkinson’s Disease. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2017, 1863, 2171–2181. [Google Scholar] [CrossRef]
- Kakhlon, O.; Breuer, W.; Munnich, A.; Cabantchik, Z.I. Iron Redistribution as a Therapeutic Strategy for Treating Diseases of Localized Iron AccumulationThis Review Is One of a Selection of Papers Published in a Special Issue on Oxidative Stress in Health and Disease. Can. J. Physiol. Pharmacol. 2010, 88, 187–196. [Google Scholar] [CrossRef]
- Tang, B.L. Sirtuins as Modifiers of Parkinson’s Disease Pathology. J. Neurosci. Res. 2017, 95, 930–942. [Google Scholar] [CrossRef]
- Mautone, N.; Zwergel, C.; Mai, A.; Rotili, D. Sirtuin Modulators: Where Are We Now? A Review of Patents from 2015 to 2019. Expert. Opin. Ther. Pat. 2020, 30, 389–407. [Google Scholar] [CrossRef]
- Anis, E.; Zafeer, M.F.; Firdaus, F.; Islam, S.N.; Anees Khan, A.; Ali, A.; Hossain, M.M. Ferulic Acid Reinstates Mitochondrial Dynamics through PGC1α Expression Modulation in 6-hydroxydopamine Lesioned Rats. Phytother. Res. 2020, 34, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Feng, D.; Tao, K.; Wang, R.; Shi, Y.; Qin, H.; Murphy, M.P.; Yang, Q.; Zhao, G. MitoQ Protects Dopaminergic Neurons in a 6-OHDA Induced PD Model by Enhancing Mfn2-Dependent Mitochondrial Fusion via Activation of PGC-1α. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 2859–2870. [Google Scholar] [CrossRef] [PubMed]
- Inose, Y.; Izumi, Y.; Takada-Takatori, Y.; Akaike, A.; Koyama, Y.; Kaneko, S.; Kume, T. Protective Effects of Nrf2–ARE Activator on Dopaminergic Neuronal Loss in Parkinson Disease Model Mice: Possible Involvement of Heme Oxygenase-1. Neurosci. Lett. 2020, 736, 135268. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Li, L.; Ying, Z.; Pan, C.; Huang, S.; Li, L.; Dai, M.; Yan, B.; Li, M.; Jiang, H.; et al. A Small Molecule That Protects the Integrity of the Electron Transfer Chain Blocks the Mitochondrial Apoptotic Pathway. Mol. Cell 2016, 63, 229–239. [Google Scholar] [CrossRef]
- Rappold, P.M.; Cui, M.; Grima, J.C.; Fan, R.Z.; de Mesy-Bentley, K.L.; Chen, L.; Zhuang, X.; Bowers, W.J.; Tieu, K. Drp1 Inhibition Attenuates Neurotoxicity and Dopamine Release Deficits in Vivo. Nat. Commun. 2014, 5, 5244. [Google Scholar] [CrossRef]
- East, D.A.; Campanella, M. Mitophagy and the Therapeutic Clearance of Damaged Mitochondria for Neuroprotection. Int. J. Biochem. Cell Biol. 2016, 79, 382–387. [Google Scholar] [CrossRef]
- Inoue, N.; Ogura, S.; Kasai, A.; Nakazawa, T.; Ikeda, K.; Higashi, S.; Isotani, A.; Baba, K.; Mochizuki, H.; Fujimura, H.; et al. Knockdown of the Mitochondria-localized Protein P13 Protects against Experimental Parkinsonism. EMBO Rep. 2018, 19, e44860. [Google Scholar] [CrossRef]
- Szelechowski, M.; Bétourné, A.; Monnet, Y.; Ferré, C.A.; Thouard, A.; Foret, C.; Peyrin, J.-M.; Hunot, S.; Gonzalez-Dunia, D. A Viral Peptide That Targets Mitochondria Protects against Neuronal Degeneration in Models of Parkinson’s Disease. Nat. Commun. 2014, 5, 5181. [Google Scholar] [CrossRef]
- Chang, J.-C.; Wu, S.-L.; Liu, K.-H.; Chen, Y.-H.; Chuang, C.-S.; Cheng, F.-C.; Su, H.-L.; Wei, Y.-H.; Kuo, S.-J.; Liu, C.-S. Allogeneic/Xenogeneic Transplantation of Peptide-Labeled Mitochondria in Parkinson’s Disease: Restoration of Mitochondria Functions and Attenuation of 6-Hydroxydopamine–Induced Neurotoxicity. Transl. Res. 2016, 170, 40–56.e3. [Google Scholar] [CrossRef]
- Pal, C. Targeting Mitochondria with Small Molecules: A Promising Strategy for Combating Parkinson’s Disease. Mitochondrion 2024, 79, 101971. [Google Scholar] [CrossRef]
- Zagare, A.; Preciat, G.; Nickels, S.L.; Luo, X.; Monzel, A.S.; Gomez-Giro, G.; Robertson, G.; Jaeger, C.; Sharif, J.; Koseki, H.; et al. Omics Data Integration Suggests a Potential Idiopathic Parkinson’s Disease Signature. Commun. Biol. 2023, 6, 1179. [Google Scholar] [CrossRef] [PubMed]
- Longoni, B.; Fasciani, I.; Kolachalam, S.; Pietrantoni, I.; Marampon, F.; Petragnano, F.; Aloisi, G.; Coppolino, M.F.; Rossi, M.; Scarselli, M.; et al. Neurotoxic and Neuroprotective Role of Exosomes in Parkinson’s Disease. Curr. Pharm. Des. 2020, 25, 4510–4522. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Wang, R.; Xu, H.; Tu, L.; Chen, H.; Li, H.; Liu, N.; Wang, J.; Li, S.; Yin, F.; et al. Identification of an Autoinhibitory, Mitophagy-Inducing Peptide Derived from the Transmembrane Domain of USP30. Autophagy 2022, 18, 2178–2197. [Google Scholar] [CrossRef] [PubMed]
- Antico, O.; Thompson, P.W.; Hertz, N.T.; Muqit, M.M.K.; Parton, L.E. Targeting Mitophagy in Neurodegenerative Diseases. Nat. Rev. Drug Discov. 2025, 24, 276–299. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Gene | Protein and Mutation Type | Shared MT Alterations | Gene-Specific MT Alterations | Age of Onset (Motor Symptoms) | Clinical Consequences |
|---|---|---|---|---|---|
| Parkin (PARK2) [27,28,29,30,31] | E3 ubiquitin ligase AR (more than 130 mutations) |
|
| <40 years | Slow progression, dystonia; infrequent olfactory dysfunction and cognitive impairment |
| PINK1 (PARK6) [27,30,31,32,33,34,35,36] | MT serine-threonine kinase AR (111 point, frameshift, and truncating mutations) |
|
| Mid-30s | Slow progression, dystonia, non -motor symptoms, occasionally psychiatric disorders |
| DJ-1 (PARK7) [37,38,39,40] | Peptidase C56 family AR (homozygous deletions or point mutations) |
|
| <50 years | Slow progression, early onset of motor symptoms, amyotrophy, cognitive impairment, acute behavioral disturbances |
| ATP13A2 (PARK9) [41,42,43,44] | Neuronal lysosomal type 5 P-type ATPase AR (loss-of-function mutations) |
|
| <20 years | Dementia, pyramidal degeneration, spasticity, supranuclear gaze palsy |
| PLA2G6 (PARK14) [3,45,46,47] | Ca2+-independent phospholipase A2β AR (more than 18 variants) |
|
| Adolescence-early 20s | Dystonia, gait impairment, speech difficulties, spasticity, myoclonus, neuropsychiatric and cognitive disorders |
| FBXO7 (PARK15) [48,49,50,51,52] | F-box proteins (FBPs) adaptor protein member AR (3 point mutations, homozygous truncating FBXO7 mutation, compound heterozygous mutations) |
|
| Childhood | Tremor, rigidity, bradykinesia, pyramidal signs |
| VPS13C (PARK23) [53,54] | Vacuolar sorting proteins 13 family AR (truncating mutations) |
|
| Early 20s | Lewy-body inclusions, cognitive decline, axial symptoms, dysautonomia |
| Gene | Protein and Mutation Type | Shared MT Alterations | Gene-Specific MT Alterations | Age of Onset (Motor Symptoms) | Clinical Consequences |
|---|---|---|---|---|---|
| SNCA (PARK1/ PARK4) [15,94,95,96,97,98] | α-synuclein AD (p.A53T, p.A30P, p.E46K, p.G51D, p.A53E, duplications, triplications) |
|
| 20–85 years | Resting tremor, bradikinesia, rigidity, dysphagia, dysarthria, cognitive deficits |
| LRRK2 (PARK8) [99,100,101,102,103,104,105,106,107,108,109] | Leucine-rich repeat kinase 2 AD (G2019S) |
|
| 30–80 years | Bradykinesia, rigidity resting tremor, gait abnormalities, postural instability, orthostatic hypotension, hallucinations, dementia, less frequent RBD and anosmia |
| Omi/HtrA2 (PARK 13) [110,111,112] | Serine protease (p.G399S and other genetic variants) |
|
| 40–70 years | Bradykinesia, muscular rigidity, tremor |
| VPS35 (PARK17) [113,114,115,116,117] | hVPS35 AD (p.D620N and other mutations) |
|
| Around 50 years | Resting tremor, rigidity, bradykinesia, postural reflexes alterations |
| CHCHD2 (PARK22) [118,119,120,121] | CHCHD2 AD (missense mutations) |
|
| Mid-50s | Early essential tremor, restless legs syndrome, depression, mild cognitive deficits |
| Gene | Protein and Mutation Type | Shared MT Alterations | Gene-Specific MT Alterations | Age of Onset (Motor Symptoms) | Clinical Consequences |
|---|---|---|---|---|---|
| ATXN3 [148,149,150,151] | Ataxin-3 protein AD (CAG repeat expansion) |
|
| Adolescence-middle age | Progressive cerebellar ataxia, pyramidal signs, dystonic-rigid extrapyramidal syndrome, peripheral amyotrophy, generalized areflexia, external ophthalmoplegia, action-induced facial and lingual fasciculations, bulging eyes |
| CLN3 [152,153] | CLN3 protein AR |
|
| Childhood (4–7 years) | Early-onset progressive vision loss, personality changes, behavioral problems, slow learning, seizures, progressive motor function loss |
| GLB1 [154] | β-galactosidase (β-gal) AR (cleavage of the terminal β-1,4-linked galactose residue from GM1 gangliosides) |
|
| Childhood-adolescence | Dystonia/hypotonia, speech difficulty, hepatosplenomegaly, developmental regression, seizures, visual impairment |
| POLG [155,156,157,158] | DNA Polymerase subunit gamma |
|
| Early childhood to third–fourth decade | Various clinical features depending on the specific syndrome |
| Toxic Agent | Toxin Type | First Identification | Mitochondrial Alterations |
|---|---|---|---|
| Rotenone [208,214,228,229,230] | Crystalline isoflavone, used as pesticide, insecticide, and piscicide | 1990s epidemiological studies in humans; first in vivo PD model in rats in 2000 |
|
| MPTP [206,207,210,231] | Tetrahydropyridine, precursor of MPP+ | Late 1970s–early 1980s toxicity found in humans (after contaminated intravenous drug use); first animal model in 1984 (squirrel monkey) |
|
| 6-OHDA [226,232] | Dopamine-derived benzenetriol | Toxicity described in 1959; first PD (akinesia) model in 1968 |
|
| Category | Agents | Mechanism of Action | Evidence Level |
|---|---|---|---|
| ETC and Antioxidants | CoQ10, Idebenone, Methylene Blue, Creatine | Bypass complex I, ETC facilitation, ROS neutralization | Preclinical and Limited Clinical |
| Phytochemicals | Resveratrol, Curcumin, Quercetin | Antioxidant activity, mitophagy, mitochondrial fusion | Preclinical |
| Bile Acids | UDCA, Taurine-UDCA | Enhances ATP, reduces ROS, stabilizes membrane potential | Preclinical |
| Metal Homeostasis | Deferiprone, Deferoxamine | Chelation of Fe, Cu; reduces oxidative stress | Preclinical |
| Peptides/Proteins | GLP-1 Agonists, SIRT1/SIRT3 | Mitochondrial biogenesis, metabolic regulation | Preclinical and Mixed Clinical |
| Signaling Modulators | MitoQ, Ferulic Acid, RNS60, TPNA10168 | Biogenesis via AMPK/PGC-1α, antioxidant response via Nrf2/ARE | Preclinical |
| Mitochondrial Dynamics | Mdivi-1, Compound A | Inhibits fission and apoptosis pathways | Preclinical |
| Mitophagy | IU1 | Induces selective clearance of dysfunctional mitochondria | Preclinical |
| Experimental Approaches | P13 Inhibition, BDV X Protein | Apoptosis regulation, mitochondrial integrity restoration | Experimental |
| Mitochondrial Transplantation | Allogeneic Mitochondria | Mitochondrial replacement therapy | Preclinical |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucchesi, M.; Biso, L.; Bonaso, M.; Longoni, B.; Buchignani, B.; Battini, R.; Santorelli, F.M.; Doccini, S.; Scarselli, M. Mitochondrial Dysfunction in Genetic and Non-Genetic Parkinson’s Disease. Int. J. Mol. Sci. 2025, 26, 4451. https://doi.org/10.3390/ijms26094451
Lucchesi M, Biso L, Bonaso M, Longoni B, Buchignani B, Battini R, Santorelli FM, Doccini S, Scarselli M. Mitochondrial Dysfunction in Genetic and Non-Genetic Parkinson’s Disease. International Journal of Molecular Sciences. 2025; 26(9):4451. https://doi.org/10.3390/ijms26094451
Chicago/Turabian StyleLucchesi, Martina, Letizia Biso, Marco Bonaso, Biancamaria Longoni, Bianca Buchignani, Roberta Battini, Filippo Maria Santorelli, Stefano Doccini, and Marco Scarselli. 2025. "Mitochondrial Dysfunction in Genetic and Non-Genetic Parkinson’s Disease" International Journal of Molecular Sciences 26, no. 9: 4451. https://doi.org/10.3390/ijms26094451
APA StyleLucchesi, M., Biso, L., Bonaso, M., Longoni, B., Buchignani, B., Battini, R., Santorelli, F. M., Doccini, S., & Scarselli, M. (2025). Mitochondrial Dysfunction in Genetic and Non-Genetic Parkinson’s Disease. International Journal of Molecular Sciences, 26(9), 4451. https://doi.org/10.3390/ijms26094451