
Radiation-Induced Synchronous Parathyroid Carcinoma and Papillary Thyroid Carcinoma: Clinical, Morphological, and Genetic Insights

 ,
,
Abstract
1. Introduction
2. Case Presentation
2.1. Medical History, Clinical Findings
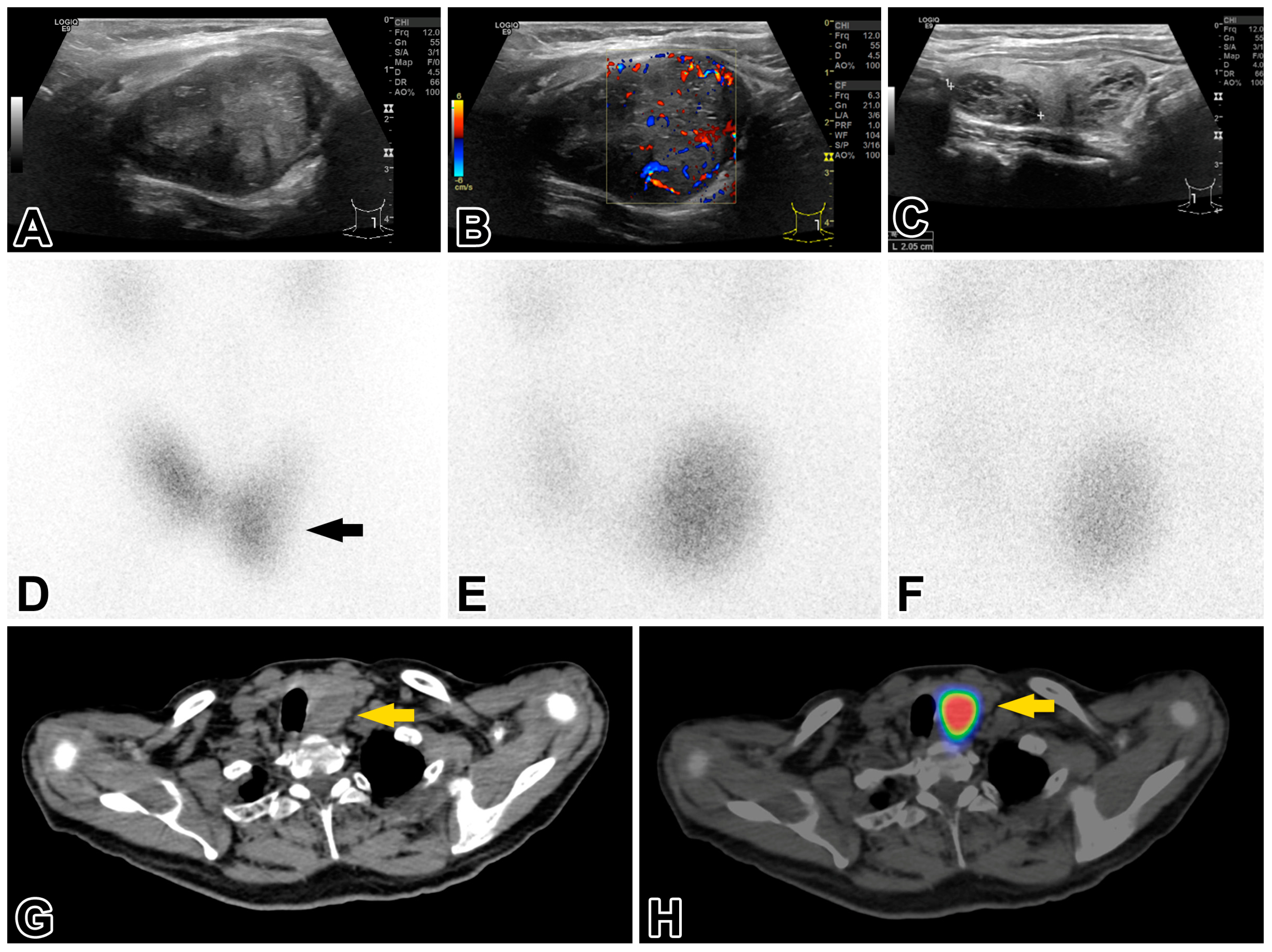
2.2. Investigations of the Thyroid and Parathyroid Gland
2.3. Pathological Investigations
2.4. Genomic Profiling of Parathyroid Carcinoma and Papillary Thyroid Carcinoma
2.5. Postoperative Treatment and Follow-Up
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roser, P.; Leca, B.M.; Coelho, C.; Schulte, K.-M.; Gilbert, J.; Drakou, E.E.; Kosmas, C.; Chuah, L.L.; Wasati, H.; Miras, A.D.; et al. Diagnosis and management of parathyroid carcinoma: A state-of-the-art review. Endocr.-Relat. Cancer. 2023, 30, e220287. [Google Scholar] [CrossRef]
- Fingeret, A.L. Contemporary evolution and management of parathyroid carcinoma. JCO Oncol. Pract. 2020, 17, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Cetani, F.; Pardi, E.; Torregrossa, L.; Borsari, S.; Pierotti, L.; Dinoi, E.; Marcocci, C. Approach to a patient with parathyroid carcinoma. J. Clin. Endocrinol. Metab. 2024, 109, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Viswanath, A.; Drakou, E.E.; Lajenunesse-Trempe, F.; Grossman, A.B.; Dimitriadis, G.K. Parathyroid carcinoma: New insights. Best Pract. Res. Clin. Endocrinol. Metab. 2025, 39, 101966. [Google Scholar] [CrossRef]
- Silva-Figueroa, A.M.; Bassett, R.; Christakis, I.; Moreno, P.; Clarke, C.N.; Busaidy, N.L.; Grubbs, E.G.; Lee, J.E.; Perrier, N.D.; Williams, M.D. Using a novel diagnostic nomogram to differentiate malignant from benign parathyroid neoplasms. Endocr. Pathol. 2019, 30, 285–296. [Google Scholar] [CrossRef]
- Juhlin, C.C.; Nilsson, I.-L.; Lagerstedt-Robinson, K.; Stenman, A.; Branström, R.; Tham, E.; Höög, A. Parafibromin immunostaining of parathyroid tumors in clinical routine: A near-decade experience from a tertiary center. Mod. Pathol. 2019, 32, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Mo, S.; Xiao, J.; Cui, M.; Zheng, Q.; Chen, T.; Chang, X. The significance of an immunohistochemical marker-based panel in assisting the diagnosis of parathyroid carcinoma. Endocrine 2024, 84, 1146–1153. [Google Scholar] [CrossRef]
- Tan, M.-H.; Morrison, C.; Wang, P.; Yang, X.; Haven, C.J.; Zhang, C.; Zhao, P.; Tretiakova, M.S.; Korpi-Hyovalti, E.; Burgess, J.R.; et al. Loss of parafibromin immunoreactivity is a distinguishing feature of parathyroid carcinoma. Clin. Cancer Res. 2004, 10, 6629–6637. [Google Scholar] [CrossRef]
- Simescu, R.; Pop, M.; Piciu, A.; Muntean, V.; Piciu, D. Association of parathyroid and differentiated thyroid carcinomas: A narrative up-to-date review of the literature. Medicina 2022, 58, 1184. [Google Scholar] [CrossRef]
- Nacef, I.B.; Khelifi, D.; Kalthoum, M.; Rojbi, I.; Riahi, I.; Mekni, S.; Ben Salah, M.; Mchirgui, N.; Khiari, K. Synchronous parathyroid carcinoma and papillary thyroid carcinoma. Clin. Case Rep. 2022, 10, e06369. [Google Scholar] [CrossRef]
- Alajaimi, A.; Altooq, N.; Chandran, N.; Alderazi, Y. Synchronous parathyroid carcinoma and nonivasive follicular thyroid neoplasm with papillary-like nuclear features. Cureus 2022, 14, e24006. [Google Scholar] [CrossRef] [PubMed]
- Marcy, P.-Y.; Thariat, J.; Sudaka, A.; Poissonet, G. Synchronous parathyroid and papillary thyroid carcinomas. Thyroid 2009, 19, 1131–1133. [Google Scholar] [CrossRef]
- Russ, G.; Bonnema, S.J.; Erdogan, M.F.; Durante, C.; Ngu, R.; Leenhardt, L. European Thyroid Association guidelines for ultrasound malignancy risk stratification of thyroid nodules in adults: The EU-TIRADS. Eur. Thyroid. J. 2017, 6, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, P.; Lu, J.; Pan, B.; Guo, D.; Zhang, Z.; Wang, M.; Sun, J.; Wang, W. Diagnostic significance of parafibromin expression in parathyroid carcinoma. Hum. Pathol. 2022, 127, 28–38. [Google Scholar] [CrossRef]
- Pearle, M.S.; Goldfarb, D.S.; Assimos, D.G.; Curhan, G.; Denu-Ciocca, C.J.; Matlaga, B.R.; Monga, M.; Penniston, K.L.; Preminger, G.M.; Turk, T.M.T.; et al. Medical management of kidney stones: AUA Guideline. J. Urol. 2014, 192, 316–324. [Google Scholar] [CrossRef] [PubMed]
- NICE Guideline—Renal and ureteric stones: Assessment and management. BJU. Int. 2019, 123, 220–232. [CrossRef]
- Osther, P.J.; Grenabo, L.; Haraldsson, G.; Holmberg, G.; Lindell, O.; Mogensen, P.; Schultz, A.; Ulvik, N.M. Metabolic evaluation and medical management of upper urinary tract stone. Scand. J. Urol. Nephrol. 1999, 33, 372–381. [Google Scholar]
- Perez, A.A.; Schneider, D.F.; Long, K.L.; Pitt, S.C.; Sippel, R. Timely evaluation and management of primary hyperparathyroidism in patients with kidney stones. J. Surg. Res. 2018, 232, 564–569. [Google Scholar] [CrossRef]
- Balakrishnan, M.; George, S.A.; Rajab, S.H.; Francis, I.M.; Kapila, K. Cytological challenges in the diagnosis of intrathyroidal carcinoma: A case report and review of the literature. Diagn. Cytopathol. 2018, 46, 47–52. [Google Scholar] [CrossRef]
- Sriphrapradang, C.; Sornmayura, P.; Chanplakorn, N.; Trachoo, O.; Sae-Chew, P.; Aroonroch, R. Fine-needle aspiration cytology of parathyroid carcinoma mimic Hürthle cell thyroid neoplasm. Case Rep. Endocrinol. 2014, 2014, 680876. [Google Scholar] [CrossRef]
- Adams, D.; Kubik, M.; Vavinskaya, V. Fine-needle aspiration of parathyroid carcinoma: A diagnostic pitfall of a rare endocrine malignancy. Am. J. Clin. Pathol. 2016, 146, S49. [Google Scholar] [CrossRef]
- Ha, H.J.; Kim, E.J.; Kim, J.-S.; Shin, M.-S.; Noh, I.; Park, S.; Koh, J.S.; Lee, S.-S. Major clues and pitfalls in the differential diagnosis of parathyroid and thyroid lesions using fine needle aspiration cytology. Medicina 2020, 56, 558. [Google Scholar] [CrossRef]
- De Falco, N.; Santangelo, G.; Chirico, F.; Cangiano, A.; Somella, M.G.; Cosenza, A.; Ronchi, A.; Accardo, M.; Pellino, G.; Parmeggiani, D.; et al. Synchronous intrathyroidal parathyroid carcinoma and thyroid carcinoma: Case report and review of the literature. BMC Endocr. Disord. 2021, 21, 60. [Google Scholar] [CrossRef] [PubMed]
- Russ, J.E.; Scanlon, E.F.; Sener, S.F. Parathyroid adenomas following irradiation. Cancer 1979, 43, 1078–1083. [Google Scholar] [CrossRef]
- McCullen, T.; Bodie, G.; Gill, A.; Ihre-Lundgreen, C.; Shun, A.; Bergin, M.; Steven, G.; Delbridge, L. Hyperparathyroidism after irradiation for childhood malignancy. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 1164–1168. [Google Scholar] [CrossRef] [PubMed]
- Christmas, T.J.; Chapple, C.R.; Noble, J.G.; Milroy, E.J.G.; Cowie, A.G.A. Hyperparathyroidism after neck irradiation. Br. J. Surg. 1988, 75, 873–874. [Google Scholar] [CrossRef]
- Veiga, L.H.S.; Holmberg, E.; Anderson, H.; Pottern, L.; Sadetzki, S.; Adams, M.J.; Sakata, R.; Schneider, A.B.; Inskip, P.; Bhatti, P. Thyroid cancer after childhood exposure to external radiation: An updated pooled analysis of 12 studies. Radiat. Res. 2016, 185, 473–484. [Google Scholar] [CrossRef]
- Tucker, M.A.; Coleman, C.N.; Cox, R.S.; Varghese, A.; Rosenberg, S.A. Risk of second malignancies following Hodgkin’s disease after 15 years. N. Eng. J. Med. 1988, 318, 76–81. [Google Scholar] [CrossRef]
- Otsuka, K.; Iwasaki, T. Insights into radiation carcinogenesis based on dose-rate effects in tissue stem cells. Int. J. Radiol. Biol. 2023, 99, 1503–1521. [Google Scholar] [CrossRef]
- Doig, K.D.; Fellowes, A.P.; Fox, S.B. Homologous recombination repair deficiency: An overview for pathologists. Mod. Pathol. 2023, 36, 100049. [Google Scholar] [CrossRef]
- Suzuki, K.; Saenko, V.; Yamashita, S.; Mitsutake, N. Radiation-induced thyroid cancers: Overview of molecular signatures. Cancers 2019, 11, 1290. [Google Scholar] [CrossRef] [PubMed]
- Chunduri, N.K.; Storchová, Z. The diverse consequences of aneuploidy. Nat. Cell Biol. 2019, 21, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Farnebo, F.; Kytöla, S.; Teh, B.T.; Dwight, T.; Wong, F.K.; Höög, A.; Elvius, M.; Wassif, W.S.; Thomson, N.W.; Farnebo, L.-O.; et al. Alternative genetic pathways in parathyroid tumorigenesis. J. Clin. Endocrinol. Metab. 1999, 84, 3775–3780. [Google Scholar] [PubMed]
- Marini, F.; Giusti, F.; Iantomasi, T.; Brandi, M.L. Parathyroid tumors: Molecular signatures. Int. J. Mol. Sci. 2021, 22, 11206. [Google Scholar] [CrossRef]
- Costa-Guda, J.; Soong, C.-P.; Parekh, V.I.; Agarwal, S.K.; Arnold, A. Germline and somatic mutations in cyclin-dependent kinase inhibitor genes CDKN1A, CDKN2B, and CDKN2C in sporadic parathyroid adenomas. Horm. Cancer 2013, 4, 301–307. [Google Scholar] [CrossRef]
- Costa-Guda, J.; Marinoni, I.; Molatore, S.; Pellegata, N.S.; Arnold, A. Somatic mutation and germline sequence abnormalities in CDKN1B, encoding p27Kip1, in sporadic parathyroid adenomas. J. Clin. Endocrinol. Metab. 2011, 96, E701–E706. [Google Scholar] [CrossRef]
- Pandya, C.; Uzilov, A.V.; Bellizzi, J.; Lau, C.Y.; Moe, A.S.; Strahl, M.; Hamou, W.; Newman, L.C.; Fink, M.Y.; Antipin, Y.; et al. Genomic profiling reveals mutational landscape in parathyroid carcinomas. JCI Insight. 2017, 2, e92061. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, X.; Wang, O.; Bi, Y.; Xing, X.; Cui, M.; Wang, M.; Tao, W.; Liao, Q.; Zhao, Y. The genomic profile of parathyroid carcinoma based on whole-genome sequencing. Int. J. Cancer. 2020, 47, 2446–2457. [Google Scholar] [CrossRef]
- Yu, W.; McPherson, J.R.; Stevenson, M.; van Eijk, R.; Heng, H.L.; Newey, P.; Gan, A.; Ruano, D.; Huang, D.; Poon, S.L.; et al. Whole-exome sequencing studies of parathyroid carcinomas reveal novel PRUNE2 mutations, distinctive mutational spectra related to APOBEC-catalyzed DNA mutagenesis and mutational enrichment in kinases associated with cell migration and invasion. J. Clin. Endocrin. Metab. 2015, 100, E360–E364. [Google Scholar] [CrossRef]
- Teleanu, M.-V.; Fuss, C.T.; Paramasivam, N.; Pirmann, S.; Mock, A.; Terkamp, C.; Kircher, S.; Landwehr, L.-S.; Lenschow, C.; Schlegel, N.; et al. Targeted therapy of advanced parathyroid carcinoma guided by genomic and transcriptomic profiling. Mol. Oncol. 2023, 17, 1343–1355. [Google Scholar] [CrossRef]
- Shattuck, T.M.; Välimäki, S.; Obara, T.; Gaz, R.D.; Clark, O.H.; Shoback, D.; Wierman, M.E.; Toyo, K.; Robbins, C.-M.; Carpten, J.D.; et al. Somatic and germ-line mutations of the HRPT2 gene in sporadic parathyroid carcinoma. N. Engl. J. Med. 2003, 349, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Pettinga, D.; Schubert, A.D.; Ladenson, P.W.; Ball, D.W.; Chung, J.H.; Schrock, A.B.; Madison, R.; Frampton, G.M.; Stephens, P.J.; et al. Genomic profiling of parathyroid carcinoma reveals genomic alterations suggesting benefit from therapy. Oncologist 2019, 24, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, A.; Masuda, M.; Tsuta, K.; Kawasaki, K.; Nakamura, Y.; Sakuma, T.; Asamura, H.; Gemma, A.; Yamada, T. Hippo pathway gene mutations in malignant mesothelioma. J. Thoracic. Oncol. 2015, 10, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, K.; Jia, Y.; Chuang, J.-C.; Sun, X.; Lin, Y.-H.; Celen, C.; Li, L.; Huang, F.; Liu, X.; et al. Dual ARID1A/ARID1B loss leads to rapid carcinogenesis and disruptive redistribution of BAF complexes. Nat. Cancer 2020, 1, 909–922. [Google Scholar] [CrossRef]
- Zhou, Y.; Qiu, C.; Fu, Q.; Li, T.; Zhang, X.; Zhu, C.; Qin, X.; Wu, B. Pan-cancer analysis of oncogenic role of RADL4L and experimental validation in hepatocellular carcinoma. J. Inflamm. Res. 2023, 16, 3997–4017. [Google Scholar] [CrossRef]
- Curia, M.C.; Catalano, T.; Aceto, G.M. MUTYH: Not just polyposis. World J. Clin. Oncol. 2020, 11, 428–449. [Google Scholar] [CrossRef]
- Chunduri, N.K.; Menges, P.; Zhang, X.; Wieland, A.; Gotsmann, V.L.; Mardin, B.R.; Buccitelli, C.; Korbel, J.O.; Willmund, F.; Kschischo, M.; et al. System approaches identify the consequences of monosomy in somatic human cells. Nat. Commun. 2021, 12, 556. [Google Scholar] [CrossRef]
- Iglesias, M.L.; Schmidt, A.; Ghuzian, A.A.; Lacroix, L.; de Vathaire, F.; Chevillard, S.; Schlumberger, M. Radiation exposure and thyroid cancer: A review. Arch. Endocrinol. Metab. 2017, 61, 180–187. [Google Scholar] [CrossRef]
- Chu, Y.-H.; Wirth, L.J.; Farahani, A.A.; Nosé, V.; Faquin, W.C.; Santagata, D.D.; Sadow, P.M. Clinicopathologic features of kinase fusion-related thyroid carcinomas: An integrative analysis with molecular characterization. Mod. Pathol. 2020, 33, 2458–2472. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Measured Value | Normal Value Range | |
|---|---|---|
| Sodium (mmol/L) | 136 | 136–145 |
| Potassium (mmol/L) | 4.8 | 3.5–5.1 |
| Calcium (mmol/L) | 3.48 | 2.20–2.55 |
| Albumin-adjusted calcium (mmol/L) | 3.46 | 2.20–2.55 |
| Magnesium (mmol/L) | 0.73 | 0.7–1.05 |
| Phosphorus (mmol/L) | 0.51 | 0.87–1.45 |
| Alkaline phosphatase (U/L) | 463 | 35–104 |
| Carbamide (mmol/L) | 6.4 | 2.1–7.1 |
| Creatinine (µmol/L) | 96 | 53–97 |
| eGFR-EPI (ml/min/1.73 m2) | 77 | 90< |
| Parathormone (pmol/L) | 150 | 1.6–6.9 |
| Urinary calcium excretion (mmol/L) | 423 | 1.25–3.75 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iványi, G.; Christofi, A.; Sipka, G.; Zombori, T.; Kuthi, L.; Simon, A.; Dobi, D.; Lázár, G.; Valkusz, Z.; Iványi, B. Radiation-Induced Synchronous Parathyroid Carcinoma and Papillary Thyroid Carcinoma: Clinical, Morphological, and Genetic Insights. Int. J. Mol. Sci. 2025, 26, 4441. https://doi.org/10.3390/ijms26094441
Iványi G, Christofi A, Sipka G, Zombori T, Kuthi L, Simon A, Dobi D, Lázár G, Valkusz Z, Iványi B. Radiation-Induced Synchronous Parathyroid Carcinoma and Papillary Thyroid Carcinoma: Clinical, Morphological, and Genetic Insights. International Journal of Molecular Sciences. 2025; 26(9):4441. https://doi.org/10.3390/ijms26094441
Chicago/Turabian StyleIványi, Gábor, Alexandros Christofi, Gábor Sipka, Tamás Zombori, Levente Kuthi, Andrea Simon, Deján Dobi, György Lázár, Zsuzsanna Valkusz, and Béla Iványi. 2025. "Radiation-Induced Synchronous Parathyroid Carcinoma and Papillary Thyroid Carcinoma: Clinical, Morphological, and Genetic Insights" International Journal of Molecular Sciences 26, no. 9: 4441. https://doi.org/10.3390/ijms26094441
APA StyleIványi, G., Christofi, A., Sipka, G., Zombori, T., Kuthi, L., Simon, A., Dobi, D., Lázár, G., Valkusz, Z., & Iványi, B. (2025). Radiation-Induced Synchronous Parathyroid Carcinoma and Papillary Thyroid Carcinoma: Clinical, Morphological, and Genetic Insights. International Journal of Molecular Sciences, 26(9), 4441. https://doi.org/10.3390/ijms26094441