
Secreted Protein Acidic and Rich in Cysteine (SPARC) Induced by the Renin–Angiotensin System Causes Endothelial Inflammation in the Early Stages of Hypertensive Vascular Injury

 , , , and
, , , and
Abstract
1. Introduction
2. Results
2.1. Physiological and Biochemical Data of Rats Treated with DOCA–Salt
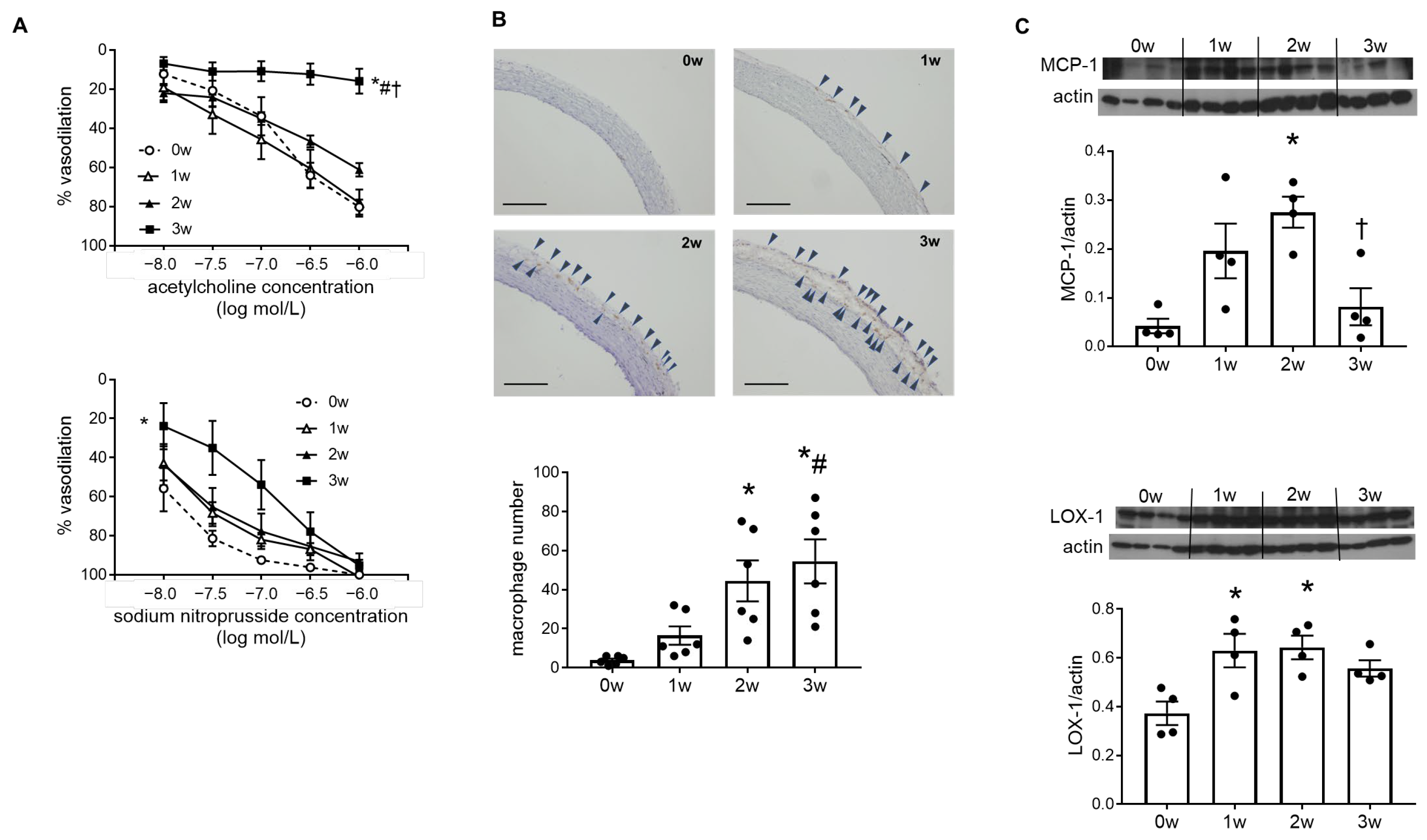
2.2. Endothelial Cell Dysfunction and Vascular Inflammation Occurred in the DOCA–Salt Rat Aorta
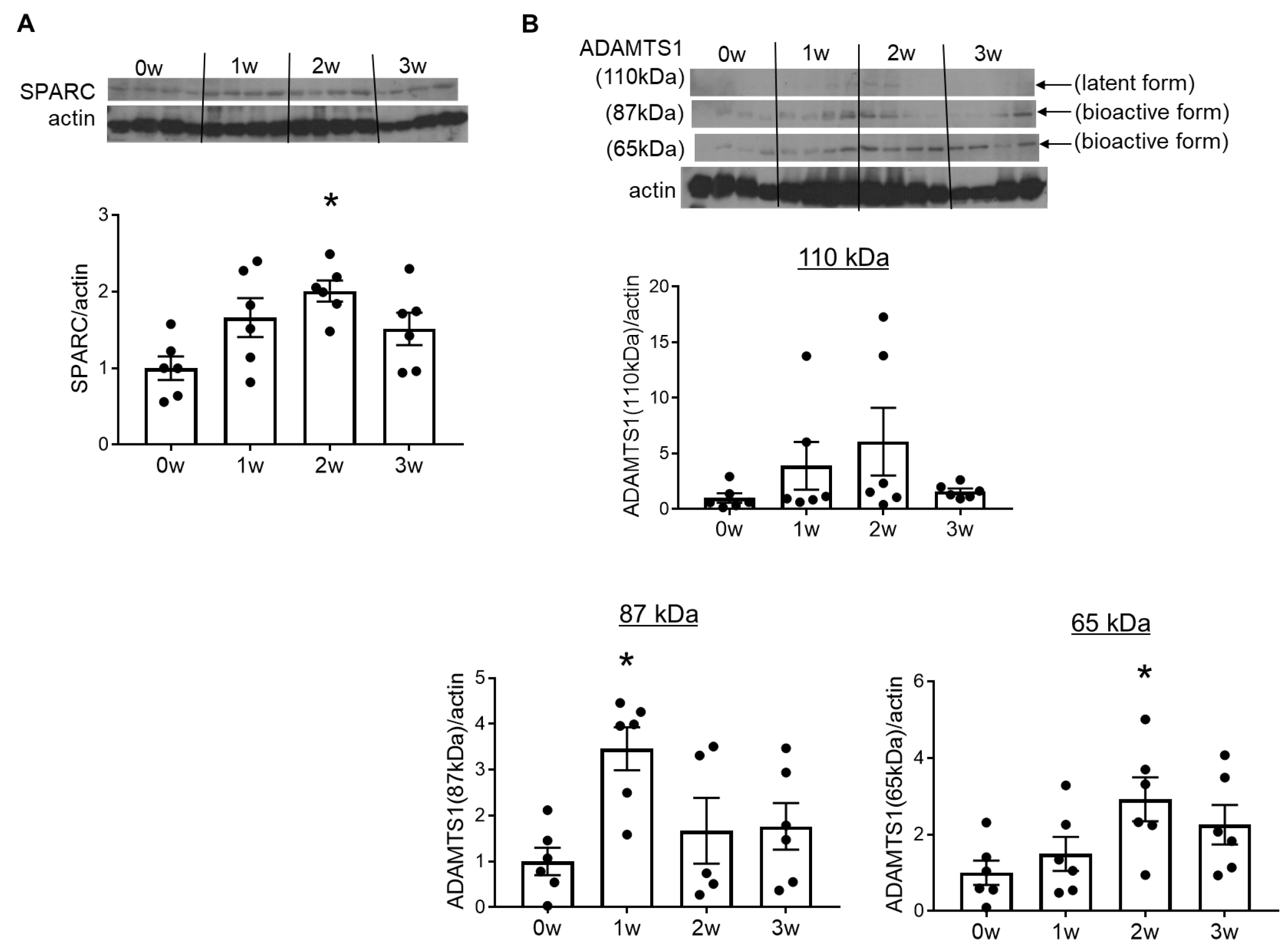
2.3. Overexpression of SPARC and ADAMTS1 Was Induced in the Early Stages of Vascular Injury in DOCA–Salt Rats
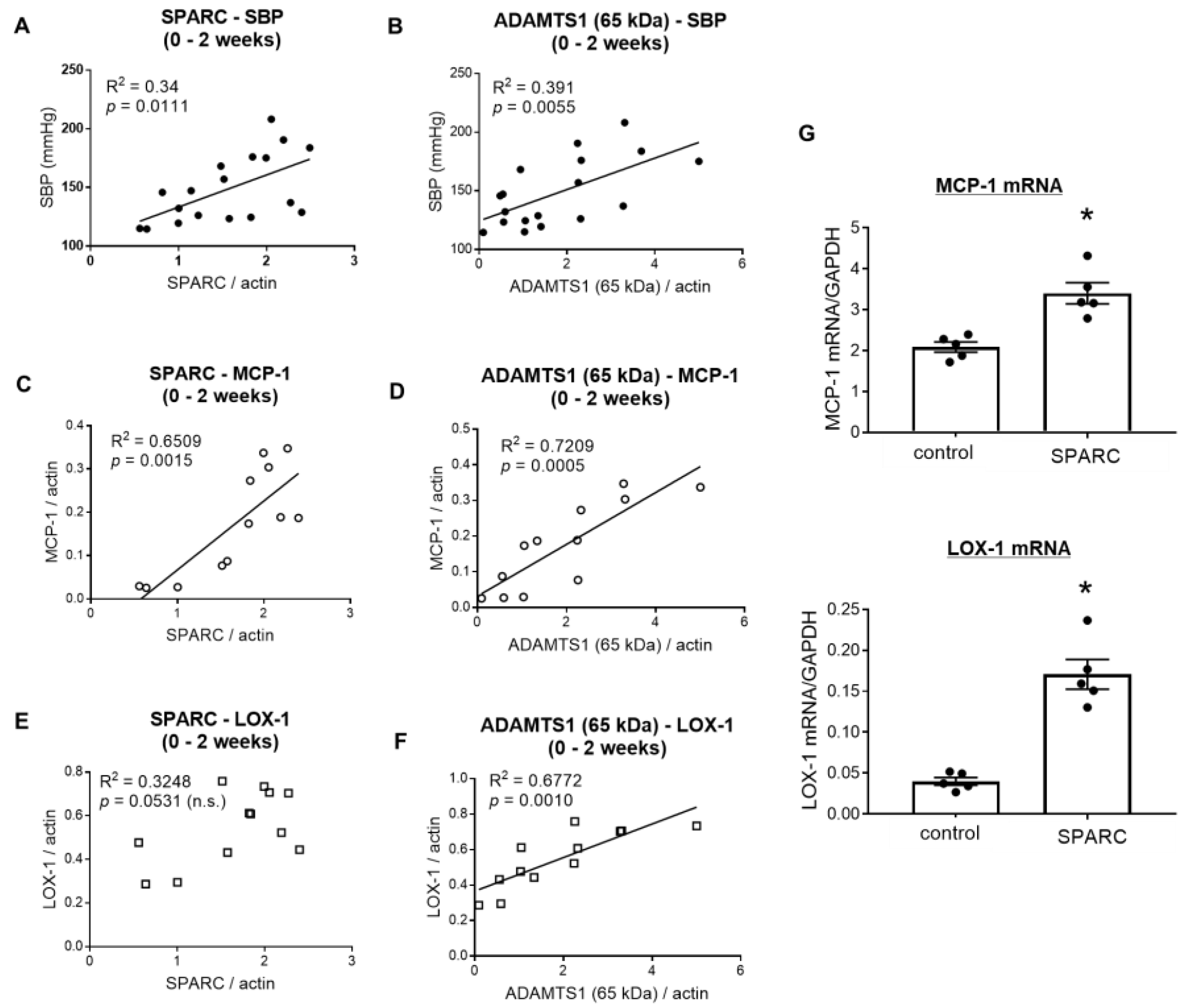
2.4. SPARC Induced Proinflammatory Gene Expression in Vascular Endothelial Cells
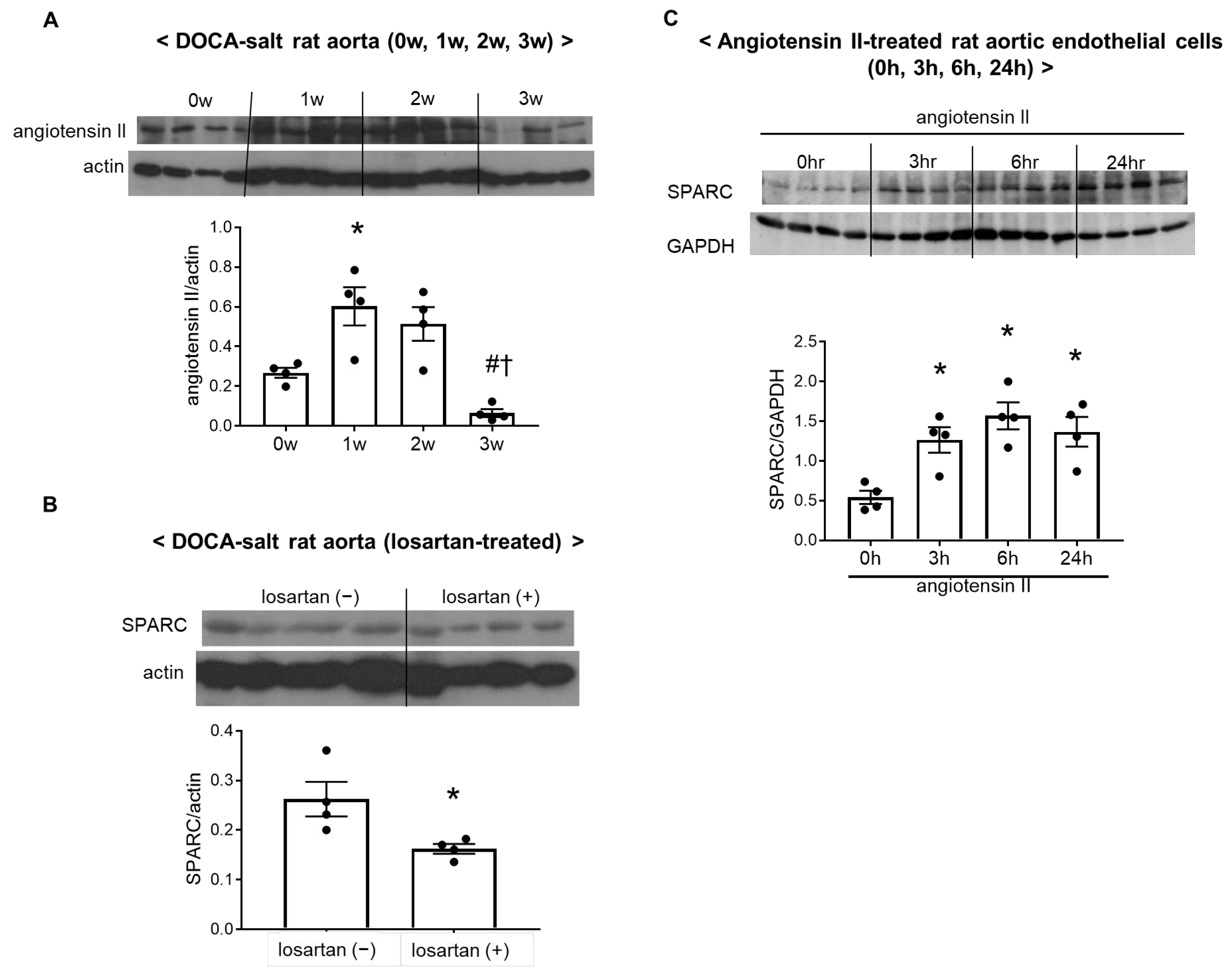
2.5. Activation of the Vascular Renin–Angiotensin System Increased SPARC Expression in DOCA–Salt Rats
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. DOCA–Salt Treatment
4.3. Urinary NOx and Plasma Malondialdehyde Levels
4.4. Vasodilatory Responses
4.5. Immunohistochemistry
4.6. Western Blotting
4.7. Cell Culture
4.8. RNA Extraction and Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poznyak, A.V.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Hypertension as a risk factor for atherosclerosis: Cardiovascular risk assessment. Front. Cardiovasc. Med. 2022, 9, 959285. [Google Scholar] [CrossRef]
- Rubanyi, G.M. The role of endothelium in cardiovascular homeostasis and diseases. J. Cardiovasc. Pharmacol. 1993, 22 (Suppl. 4), S1–S14. [Google Scholar] [CrossRef]
- Garcia-Palmieri, M.R. The endothelium in health and in cardiovascular disease. P. R. Health Sci. J. 1997, 16, 136–141. [Google Scholar]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Brekken, R.A.; Sage, E.H. SPARC, a matricellular protein: At the crossroads of cell-matrix. Matrix Biol. J. Int. Soc. Matrix Biol. 2000, 19, 569–580. [Google Scholar] [CrossRef]
- Sage, E.H.; Bornstein, P. Extracellular proteins that modulate cell-matrix interactions. SPARC, tenascin, and thrombospondin. J. Biol. Chem. 1991, 266, 14831–14834. [Google Scholar] [CrossRef]
- Toba, H.; de Castro Bras, L.E.; Baicu, C.F.; Zile, M.R.; Lindsey, M.L.; Bradshaw, A.D. Secreted protein acidic and rich in cysteine facilitates age-related cardiac inflammation and macrophage M1 polarization. Am. J. Physiol. Cell Physiol. 2015, 308, C972–C982. [Google Scholar] [CrossRef]
- Ng, Y.L.; Klopcic, B.; Lloyd, F.; Forrest, C.; Greene, W.; Lawrance, I.C. Secreted protein acidic and rich in cysteine (SPARC) exacerbates colonic inflammatory symptoms in dextran sodium sulphate-induced murine colitis. PLoS ONE 2013, 8, e77575. [Google Scholar] [CrossRef]
- Socha, M.J.; Manhiani, M.; Said, N.; Imig, J.D.; Motamed, K. Secreted protein acidic and rich in cysteine deficiency ameliorates renal inflammation and fibrosis in angiotensin hypertension. Am. J. Pathol. 2007, 171, 1104–1112. [Google Scholar] [CrossRef]
- Rempel, S.A.; Hawley, R.C.; Gutierrez, J.A.; Mouzon, E.; Bobbitt, K.R.; Lemke, N.; Schultz, C.R.; Schultz, L.R.; Golembieski, W.; Koblinski, J.; et al. Splenic and immune alterations of the Sparc-null mouse accompany a lack of immune response. Genes. Immun. 2007, 8, 262–274. [Google Scholar] [CrossRef]
- Xu, L.; Ping, F.; Yin, J.; Xiao, X.; Xiang, H.; Ballantyne, C.M.; Wu, H.; Li, M. Elevated plasma SPARC levels are associated with insulin resistance, dyslipidemia, and inflammation in gestational diabetes mellitus. PLoS ONE 2013, 8, e81615. [Google Scholar] [CrossRef]
- Toba, H.; Ikemoto, M.J.; Kobara, M.; Nakata, T. Secreted protein acidic and rich in cysteine (SPARC) and a disintegrin and metalloproteinase with thrombospondin type 1 motif (ADAMTS1) increments by the renin-angiotensin system induce renal fibrosis in deoxycorticosterone acetate-salt hypertensive rats. Eur. J. Pharmacol. 2022, 914, 174681. [Google Scholar] [CrossRef]
- Matsusaka, T.; Ichikawa, I. Biological functions of angiotensin and its receptors. Annu. Rev. Physiol. 1997, 59, 395–412. [Google Scholar] [CrossRef]
- Dzau, V.; Braunwald, E. Resolved and unresolved issues in the prevention and treatment of coronary artery disease: A workshop consensus statement. Am. Heart J. 1991, 121, 1244–1263. [Google Scholar] [CrossRef]
- Toba, H.; de Castro Bras, L.E.; Baicu, C.F.; Zile, M.R.; Lindsey, M.L.; Bradshaw, A.D. Increased ADAMTS1 mediates SPARC-dependent collagen deposition in the aging myocardium. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E1027–E1035. [Google Scholar] [CrossRef]
- Toba, H.; Jin, D.; Takai, S. Suppressing SPARC gene with siRNA exerts therapeutic effects and inhibits MMP-2/9 and ADAMTS1 overexpression in a murine model of ischemia/reperfusion-induced acute kidney injury. J. Pharmacol. Sci. 2025, 158, 103–112. [Google Scholar] [CrossRef]
- Nakashima, Y.; Raines, E.W.; Plump, A.S.; Breslow, J.L.; Ross, R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 842–851. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Chen, M.; Masaki, T.; Sawamura, T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: Implications in endothelial dysfunction and atherosclerosis. Pharmacol. Ther. 2002, 95, 89–100. [Google Scholar] [CrossRef]
- Rodriguez-Manzaneque, J.C.; Milchanowski, A.B.; Dufour, E.K.; Leduc, R.; Iruela-Arispe, M.L. Characterization of METH-1/ADAMTS1 processing reveals two distinct active forms. J. Biol. Chem. 2000, 275, 33471–33479. [Google Scholar] [CrossRef]
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar] [CrossRef]
- Yoshimoto, R.; Fujita, Y.; Kakino, A.; Iwamoto, S.; Takaya, T.; Sawamura, T. The discovery of LOX-1, its ligands and clinical significance. Cardiovasc. Drugs Ther. 2011, 25, 379–391. [Google Scholar] [CrossRef]
- Li, X.; Zhao, W.; Li, X.; Chen, X.; Li, Y.; He, J.; Qin, Y.; Li, L.; Zhang, H. The association of SPARC with hypertension and its function in endothelial-dependent relaxation. Atherosclerosis 2024, 388, 117390. [Google Scholar] [CrossRef]
- Veith, C.; Varturk-Ozcan, I.; Wujak, M.; Hadzic, S.; Wu, C.Y.; Knoepp, F.; Kraut, S.; Petrovic, A.; Gredic, M.; Pak, O.; et al. SPARC, a Novel Regulator of Vascular Cell Function in Pulmonary Hypertension. Circulation 2022, 145, 916–933. [Google Scholar] [CrossRef]
- Qi, D.; Hu, X.; Wu, X.; Merk, M.; Leng, L.; Bucala, R.; Young, L.H. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J. Clin. Investig. 2009, 119, 3807–3816. [Google Scholar] [CrossRef]
- Workman, G.; Sage, E.H. Identification of a sequence in the matricellular protein SPARC that interacts with the scavenger receptor stabilin-1. J. Cell. Biochem. 2011, 112, 1003–1008. [Google Scholar] [CrossRef]
- Okada, T.; Suzuki, H.; Travis, Z.D.; Altay, O.; Tang, J.; Zhang, J.H. SPARC Aggravates Blood-Brain Barrier Disruption via Integrin alphaVbeta3/MAPKs/MMP-9 Signaling Pathway after Subarachnoid Hemorrhage. Oxid. Med. Cell Longev. 2021, 2021, 9739977. [Google Scholar] [CrossRef]
- Koepke, J.P.; Jones, S.; DiBona, G.F. Renal nerve activity and renal function during environmental stress in DOCA-NaCl rats. Am. J. Physiol. 1986, 251, R289–R294. [Google Scholar] [CrossRef]
- Shimamura, T. 11-Deoxycorticosterone-induced hypertension, glomerulosclerosis and renal arterial and arteriolar lesions. Jpn. J. Exp. Med. 1988, 58, 225–228. [Google Scholar]
- Michel, B.; Grima, M.; Stephan, D.; Coquard, C.; Welsch, C.; Barthelmebs, M.; Imbs, J.L. Plasma renin activity and changes in tissue angiotensin converting enzyme. J. Hypertens. 1994, 12, 577–584. [Google Scholar] [CrossRef]
- Wada, T.; Kanagawa, R.; Ishimura, Y.; Inada, Y.; Nishikawa, K. Role of angiotensin II in cerebrovascular and renal damage in deoxycorticosterone acetate-salt hypertensive rats. J. Hypertens. 1995, 13, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.; Cox, A.; Roe, C.J.; Dziadek, M.; Cooper, M.E.; Gilbert, R.E. Secreted protein acidic and rich in cysteine expression after subtotal nephrectomy and blockade of the renin-angiotensin system. J. Am. Soc. Nephrol. 1997, 8, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Toba, H.; Tojo, C.; Wang, J.; Noda, K.; Kobara, M.; Nakata, T. Telmisartan inhibits vascular dysfunction and inflammation via activation of peroxisome proliferator-activated receptor-gamma in subtotal nephrectomized rat. Eur. J. Pharmacol. 2012, 685, 91–98. [Google Scholar] [CrossRef]
- Toba, H.; Wang, J.; Ohigashi, M.; Kobara, M.; Nakata, T. Telmisartan protects against vascular dysfunction with peroxisome proliferator-activated receptor-gamma activation in hypertensive 5/6 nephrectomized rats. Pharmacology 2013, 92, 265–275. [Google Scholar] [CrossRef]
- Malaval, L.; Ffrench, M.; Delmas, P.D. Circulating levels of osteonectin in normal subjects and patients with thrombocytopenia. Bone Miner. 1990, 9, 129–135. [Google Scholar] [CrossRef]
- Malaval, L.; Fournier, B.; Delmas, P.D. Radioimmunoassay for osteonectin. Concentrations in bone, nonmineralized tissues, and blood. J. Bone Miner. Res. 1987, 2, 457–465. [Google Scholar] [CrossRef]
- Serebruany, V.L.; Murugesan, S.R.; Pothula, A.; Atar, D.; Lowry, D.R.; O’Connor, C.M.; Gurbel, P.A. Increased soluble platelet/endothelial cellular adhesion molecule-1 and osteonectin levels in patients with severe congestive heart failure. Independence of disease etiology, and antecedent aspirin therapy. Eur. J. Heart Fail. 1999, 1, 243–249. [Google Scholar] [CrossRef]
- Sandy, J.D.; Westling, J.; Kenagy, R.D.; Iruela-Arispe, M.L.; Verscharen, C.; Rodriguez-Mazaneque, J.C.; Zimmermann, D.R.; Lemire, J.M.; Fischer, J.W.; Wight, T.N.; et al. Versican V1 proteolysis in human aorta in vivo occurs at the Glu441-Ala442 bond, a site that is cleaved by recombinant ADAMTS-1 and ADAMTS-4. J. Biol. Chem. 2001, 276, 13372–13378. [Google Scholar] [CrossRef]
- Jonsson-Rylander, A.C.; Nilsson, T.; Fritsche-Danielson, R.; Hammarstrom, A.; Behrendt, M.; Andersson, J.O.; Lindgren, K.; Andersson, A.K.; Wallbrandt, P.; Rosengren, B.; et al. Role of ADAMTS-1 in atherosclerosis: Remodeling of carotid artery, immunohistochemistry, and proteolysis of versican. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 180–185. [Google Scholar] [CrossRef]
- Okamoto, K.; Aoki, K. Development of a strain of spontaneously hypertensive rats. Jpn. Circ. J. 1963, 27, 282–293. [Google Scholar] [CrossRef]
- Bourd-Boittin, K.; Bonnier, D.; Leyme, A.; Mari, B.; Tuffery, P.; Samson, M.; Ezan, F.; Baffet, G.; Theret, N. Protease profiling of liver fibrosis reveals the ADAM metallopeptidase with thrombospondin type 1 motif, 1 as a central activator of transforming growth factor beta. Hepatology 2011, 54, 2173–2184. [Google Scholar] [CrossRef] [PubMed]
- Francki, A.; Bradshaw, A.D.; Bassuk, J.A.; Howe, C.C.; Couser, W.G.; Sage, E.H. SPARC regulates the expression of collagen type I and transforming growth factor-beta1 in mesangial cells. J. Biol. Chem. 1999, 274, 32145–32152. [Google Scholar] [CrossRef] [PubMed]
- Pichler, R.H.; Hugo, C.; Shankland, S.J.; Reed, M.J.; Bassuk, J.A.; Andoh, T.F.; Lombardi, D.M.; Schwartz, S.M.; Bennett, W.M.; Alpers, C.E.; et al. SPARC is expressed in renal interstitial fibrosis and in renal vascular injury. Kidney Int. 1996, 50, 1978–1989. [Google Scholar] [CrossRef] [PubMed]
- Toba, H.; Morishita, M.; Tojo, C.; Nakano, A.; Oshima, Y.; Kojima, Y.; Yoshida, M.; Nakashima, K.; Wang, J.; Kobara, M.; et al. Recombinant human erythropoietin ameliorated endothelial dysfunction and macrophage infiltration by increasing nitric oxide in hypertensive 5/6 nephrectomized rat aorta. Eur. J. Pharmacol. 2011, 656, 81–87. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0w | 1w | 2w | 3w | |
|---|---|---|---|---|
| Body weight (g) | 180 ± 4.6 | 220 ± 4.9 * | 226 ± 8.0 * | 218 ± 6.5 * |
| SBP (mmHg) | 119 ± 2.4 | 139 ± 2.6 * | 176 ± 4.1 *# | 191 ± 4.6 *#† |
| HR (bpm) | 438 ± 7.3 | 403 ± 7.2 * | 403 ± 5.9 * | 420 ± 12 |
| Urinary NOx (μmol/L) | 4.79 ± 0.85 | 4.63 ± 0.55 | 2.85 ± 0.61 | 1.70 ± 0.26 *# |
| Plasma MDA (μmol/L) | 17.7 ± 1.2 | 17.6 ± 1.5 | 17.6 ± 1.3 | 21.9 ± 2.1 |
| Gene Name | Primer Sequences |
|---|---|
| MCP-1 | forward → 5′-TGTTCAGCATTGCTGCCTGT-3′ |
| reverse → 5′-GATCTCACTTGGTTCTGGTC-3′ | |
| LOX-1 | forward → 5′-CTCAACTGGAAGCTGAATGG-3′ |
| reverse → 5′-GGTGGAATGGGAAGTTGCTT-3′ | |
| GAPDH | forward → 5′-GGCACAGTCAAGGCTGAGAATG-3′ |
| reverse → 5′-ATGGTGGTGAAGACGCCAGTA-3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toba, H.; Ikemoto, M.J.; Kobara, M.; Jin, D.; Takai, S.; Nakata, T. Secreted Protein Acidic and Rich in Cysteine (SPARC) Induced by the Renin–Angiotensin System Causes Endothelial Inflammation in the Early Stages of Hypertensive Vascular Injury. Int. J. Mol. Sci. 2025, 26, 4414. https://doi.org/10.3390/ijms26094414
Toba H, Ikemoto MJ, Kobara M, Jin D, Takai S, Nakata T. Secreted Protein Acidic and Rich in Cysteine (SPARC) Induced by the Renin–Angiotensin System Causes Endothelial Inflammation in the Early Stages of Hypertensive Vascular Injury. International Journal of Molecular Sciences. 2025; 26(9):4414. https://doi.org/10.3390/ijms26094414
Chicago/Turabian StyleToba, Hiroe, Mitsushi J. Ikemoto, Miyuki Kobara, Denan Jin, Shinji Takai, and Tetsuo Nakata. 2025. "Secreted Protein Acidic and Rich in Cysteine (SPARC) Induced by the Renin–Angiotensin System Causes Endothelial Inflammation in the Early Stages of Hypertensive Vascular Injury" International Journal of Molecular Sciences 26, no. 9: 4414. https://doi.org/10.3390/ijms26094414
APA StyleToba, H., Ikemoto, M. J., Kobara, M., Jin, D., Takai, S., & Nakata, T. (2025). Secreted Protein Acidic and Rich in Cysteine (SPARC) Induced by the Renin–Angiotensin System Causes Endothelial Inflammation in the Early Stages of Hypertensive Vascular Injury. International Journal of Molecular Sciences, 26(9), 4414. https://doi.org/10.3390/ijms26094414