
Potential Pathogenetic Role of the D313Y Mutation in the GLA Gene in Anderson Fabry Disease: Two Case Reports

 ,
,  ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
2. Detailed Case Descriptions
2.1. Methodology
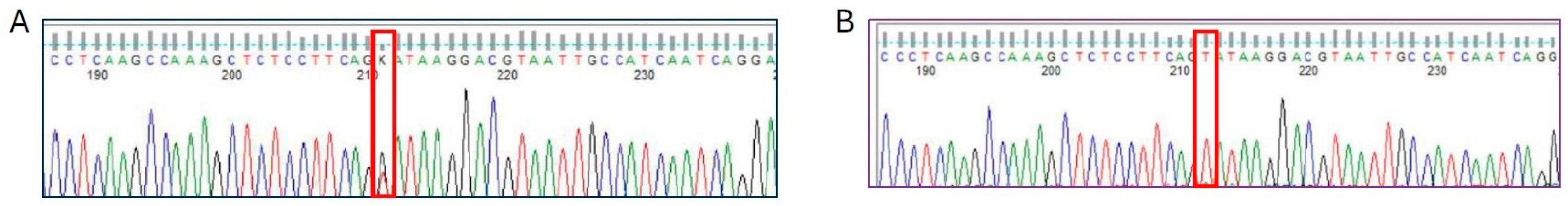
2.2. Molecular Findings
2.3. Characteristics of the Patients
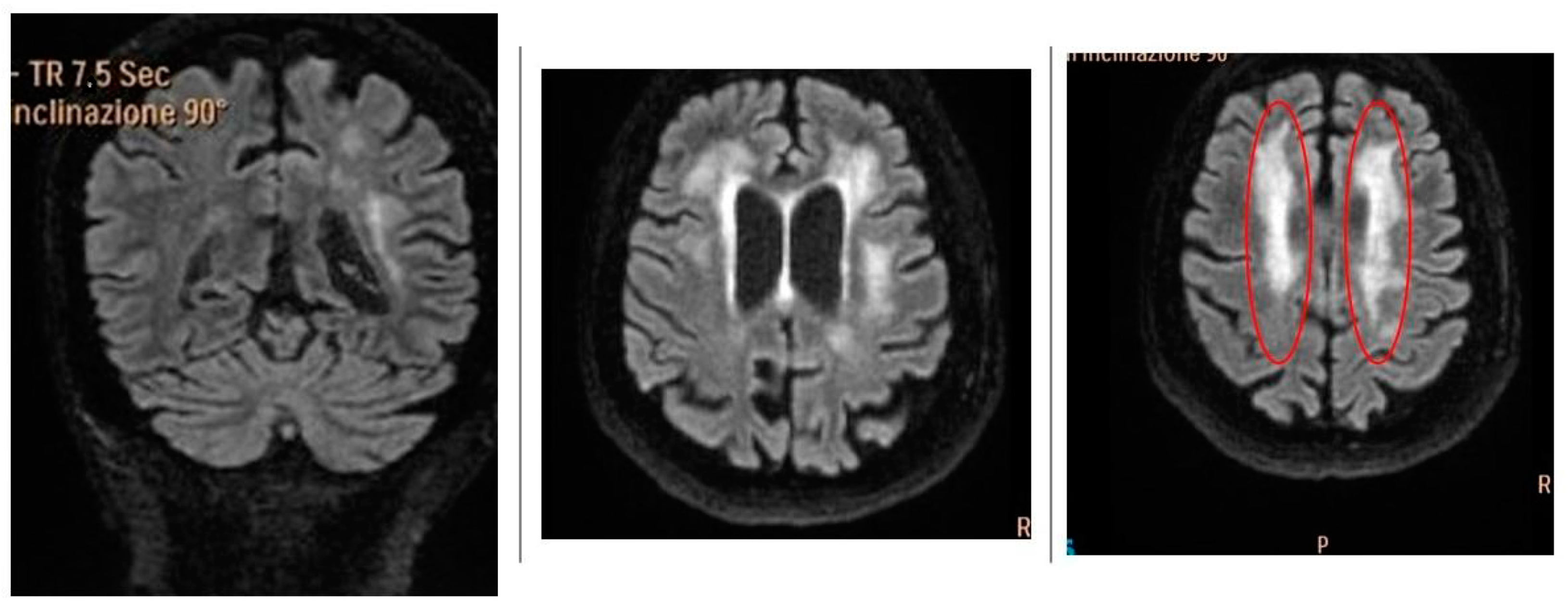
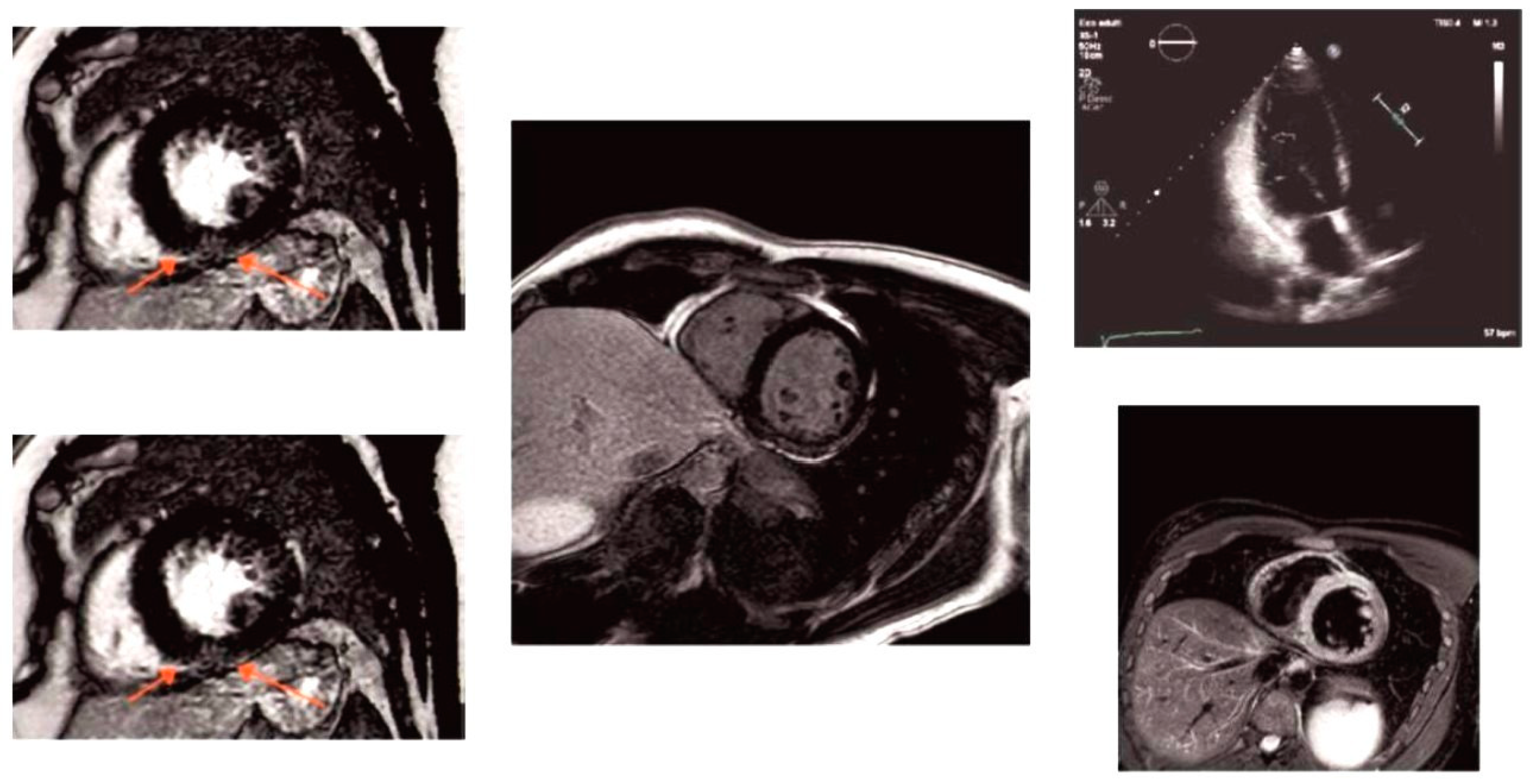
2.3.1. Patient 1
2.3.2. Patient 2
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| α-Gal A | α-galactosidase A |
| VUS | Variant of unknown significance |
| UTR | 5′ untranslated region |
| MRI | Magnetic resonance imaging |
| CSF | Cerebrospinal fluid |
| WMLs | White matter lesions |
| NSAIDs | Non-Steroidal Anti-Inflammatory Drugs |
| XCI | X-chromosome inactivation |
| TFEB | Transcription Factor EB |
| CLEAR | Coordinated Lysosomal Expression and Regulator |
| CADASIL | Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy |
References
- Germain, D.P.; Altarescu, G.; Barriales-Villa, R.; Mignani, R.; Pawlaczyk, K.; Pieruzzi, F.; Terryn, W.; Vujkovac, B.; Ortiz, A. An Expert Consensus on Practical Clinical Recommendations and Guidance for Patients with Classic Fabry Disease. Mol. Genet. Metab. 2022, 137, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Germain, D.P. Fabry Disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Cammisa, M.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. The Large Phenotypic Spectrum of Fabry Disease Requires Graduated Diagnosis and Personalized Therapy: A Meta-Analysis Can Help to Differentiate Missense Mutations. Int. J. Mol. Sci. 2016, 17, 2010. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Koulousios, K.; Stylianou, K.; Pateinakis, P.; Zamanakou, M.; Loules, G.; Manou, E.; Kyriklidou, P.; Katsinas, C.; Ouzouni, A.; Kyriazis, J.; et al. Fabry Disease Due to D313Y and Novel GLA Mutations. BMJ Open 2017, 7, e017098. [Google Scholar] [CrossRef] [PubMed]
- Palaiodimou, L.; Stefanou, M.-I.; Bakola, E.; Papadopoulou, M.; Kokotis, P.; Vrettou, A.-R.; Kapsia, E.; Petras, D.; Anastasakis, A.; Xifaras, N.; et al. D313Y Variant in Fabry Disease: A Systematic Review and Meta-Analysis. Neurology 2022, 99, e2188–e2200. [Google Scholar] [CrossRef] [PubMed]
- Consolato, F.; De Fusco, M.; Schaeffer, C.; Pieruzzi, F.; Scolari, F.; Gallieni, M.; Lanzani, C.; Feriozzi, S.; Rampoldi, L. α-Gal A Missense Variants Associated with Fabry Disease Can Lead to ER Stress and Induction of the Unfolded Protein Response. Mol. Genet. Metab. Rep. 2022, 33, 100926. [Google Scholar] [CrossRef] [PubMed]
- Gervas-Arruga, J.; Cebolla, J.J.; Irun, P.; Perez-Lopez, J.; Plaza, L.; Roche, J.C.; Capablo, J.L.; Rodriguez-Rey, J.C.; Pocovi, M.; Giraldo, P. Increased Glycolipid Storage Produced by the Inheritance of a Complex Intronic Haplotype in the α-Galactosidase A (GLA) Gene. BMC Genet. 2015, 16, 109. [Google Scholar] [CrossRef]
- Schelleckes, M.; Lenders, M.; Guske, K.; Schmitz, B.; Tanislav, C.; Ständer, S.; Metze, D.; Katona, I.; Weis, J.; Brand, S.-M.; et al. Cryptogenic Stroke and Small Fiber Neuropathy of Unknown Etiology in Patients with Alpha-Galactosidase A-10T Genotype. Orphanet J. Rare Dis. 2014, 9, 178. [Google Scholar] [CrossRef]
- Bei, E.; Antonopoulos, A.S.; Tsivgoulis, G.; Vlachopoulos, C. Fabry Disease Due to D313Y Variant with Renal Failure and Possible Cardiac Involvement: A Case Report. Eur. Heart J. Case Rep. 2023, 7, ytad224. [Google Scholar] [CrossRef] [PubMed]
- Santostefano, M.; Cappuccilli, M.; Gibertoni, D.; Fabbrizio, B.; Malvi, D.; Demetri, M.; Capelli, I.; Tringali, E.; Papa, V.; Biagini, E.; et al. Fabry Disease Nephropathy: Histological Changes with Nonclassical Mutations and Genetic Variants of Unknown Significance. Am. J. Kidney Dis. 2023, 82, 581–596.e0. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Duning, T.; Schelleckes, M.; Schmitz, B.; Stander, S.; Rolfs, A.; Brand, S.-M.; Brand, E. Multifocal White Matter Lesions Associated with the D313Y Mutation of the α-Galactosidase A Gene. PLoS ONE 2013, 8, e55565. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolny, R.; Dvorakova, L.; Ledvinova, J.; Magage, S.; Bultas, J.; Lubanda, J.C.; Elleder, M.; Karetova, D.; Pavlikova, M.; Hrebicek, M. Relationship between X-Inactivation and Clinical Involvement in Fabry Heterozygotes. Eleven Novel Mutations in the Alpha-Galactosidase A Gene in the Czech and Slovak Population. J. Mol. Med. 2005, 83, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Izhar, R.; Borriello, M.; La Russa, A.; Di Paola, R.; De, A.; Capasso, G.; Ingrosso, D.; Perna, A.F.; Simeoni, M. Fabry Disease in Women: Genetic Basis, Available Biomarkers, and Clinical Manifestations. Genes 2023, 15, 37. [Google Scholar] [CrossRef] [PubMed]
- Alfen, F.; Putscher, E.; Hecker, M.; Zettl, U.K.; Hermann, A.; Lukas, J. Abnormal Pre-mRNA Splicing in Exonic Fabry Disease-Causing GLA Mutations. Int. J. Mol. Sci. 2022, 23, 15261. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB Links Autophagy to Lysosomal Biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed]
- Franco-Juárez, B.; Coronel-Cruz, C.; Hernández-Ochoa, B.; Gómez-Manzo, S.; Cárdenas-Rodríguez, N.; Arreguin-Espinosa, R.; Bandala, C.; Canseco-Ávila, L.M.; Ortega-Cuellar, D. TFEB; Beyond Its Role as an Autophagy and Lysosomes Regulator. Cells 2022, 11, 3153. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Sample/Applications | Diagnostic/Risk Factors | Results | |
|---|---|---|---|
| Neurological | Cerebrospinal fluid |
| Normal |
| Cerebral MR angiography Catheter angiography | Cerebral vasculitis | Normal | |
| Cardiac | ECG | Sinus bradyarrhythmia | |
| Renal | Serum | eGFP++, proteinuria/albuminuria | Normal |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Russa, A.; Siniscalchi, A.; Bonaventura, A.; Di Noia, D.; Valsania, T.; Stallone, G.; Tartaglia, L.; Chiapparino, C.; Di Rienzo, G.; Coppolino, G.; et al. Potential Pathogenetic Role of the D313Y Mutation in the GLA Gene in Anderson Fabry Disease: Two Case Reports. Int. J. Mol. Sci. 2025, 26, 4400. https://doi.org/10.3390/ijms26094400
La Russa A, Siniscalchi A, Bonaventura A, Di Noia D, Valsania T, Stallone G, Tartaglia L, Chiapparino C, Di Rienzo G, Coppolino G, et al. Potential Pathogenetic Role of the D313Y Mutation in the GLA Gene in Anderson Fabry Disease: Two Case Reports. International Journal of Molecular Sciences. 2025; 26(9):4400. https://doi.org/10.3390/ijms26094400
Chicago/Turabian StyleLa Russa, Antonella, Antonio Siniscalchi, Ardito Bonaventura, Domenico Di Noia, Teresa Valsania, Giovanni Stallone, Luciano Tartaglia, Concetta Chiapparino, Giovanni Di Rienzo, Giuseppe Coppolino, and et al. 2025. "Potential Pathogenetic Role of the D313Y Mutation in the GLA Gene in Anderson Fabry Disease: Two Case Reports" International Journal of Molecular Sciences 26, no. 9: 4400. https://doi.org/10.3390/ijms26094400
APA StyleLa Russa, A., Siniscalchi, A., Bonaventura, A., Di Noia, D., Valsania, T., Stallone, G., Tartaglia, L., Chiapparino, C., Di Rienzo, G., Coppolino, G., Bolignano, D., Faga, T., Michael, A., Montesanto, A., Serra, R., & Andreucci, M. (2025). Potential Pathogenetic Role of the D313Y Mutation in the GLA Gene in Anderson Fabry Disease: Two Case Reports. International Journal of Molecular Sciences, 26(9), 4400. https://doi.org/10.3390/ijms26094400