
CRISPR/nCas9-Edited CD34+ Cells Rescue Mucopolysaccharidosis IVA Fibroblasts Phenotype

 ,
,  ,
,  and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
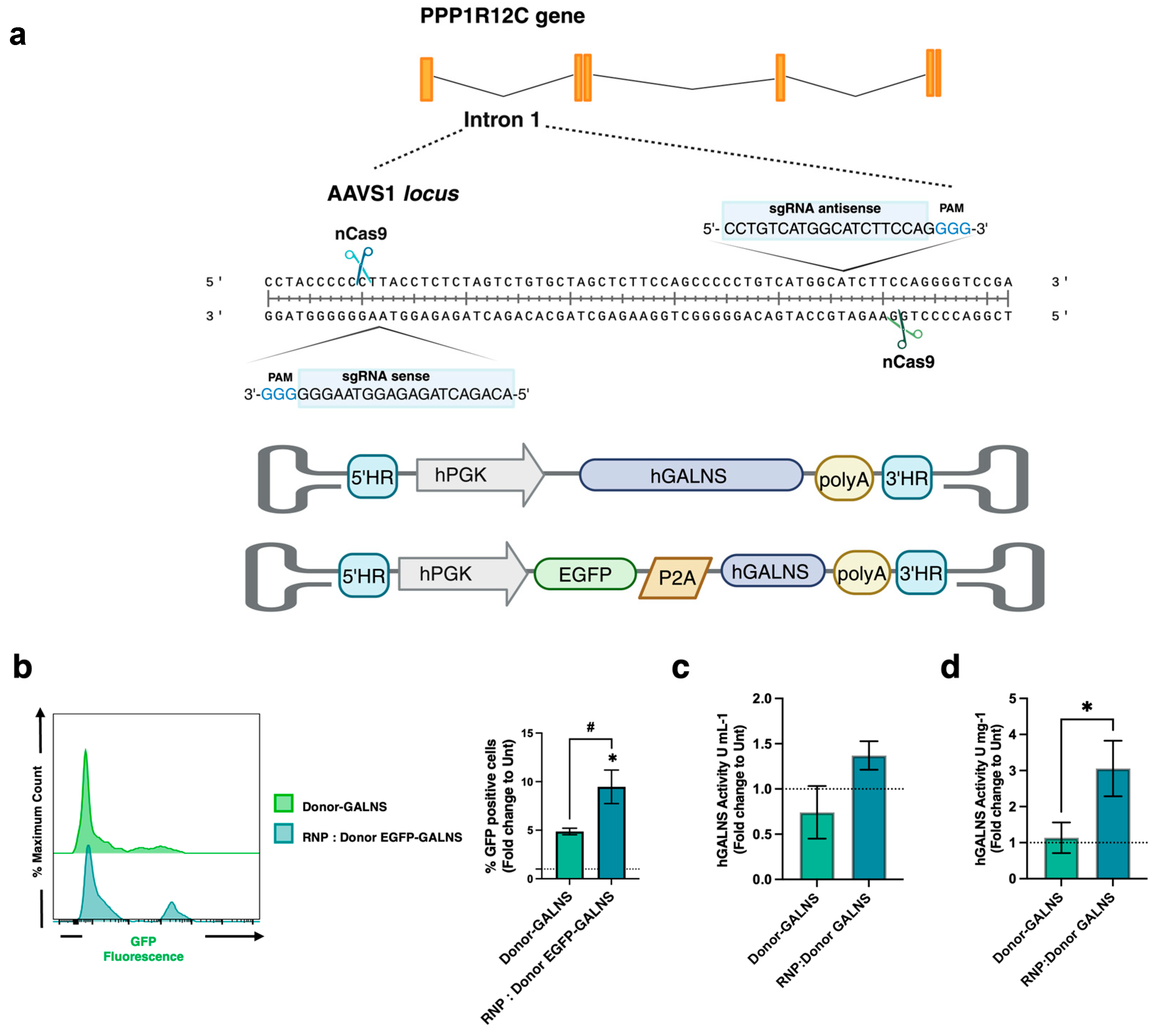
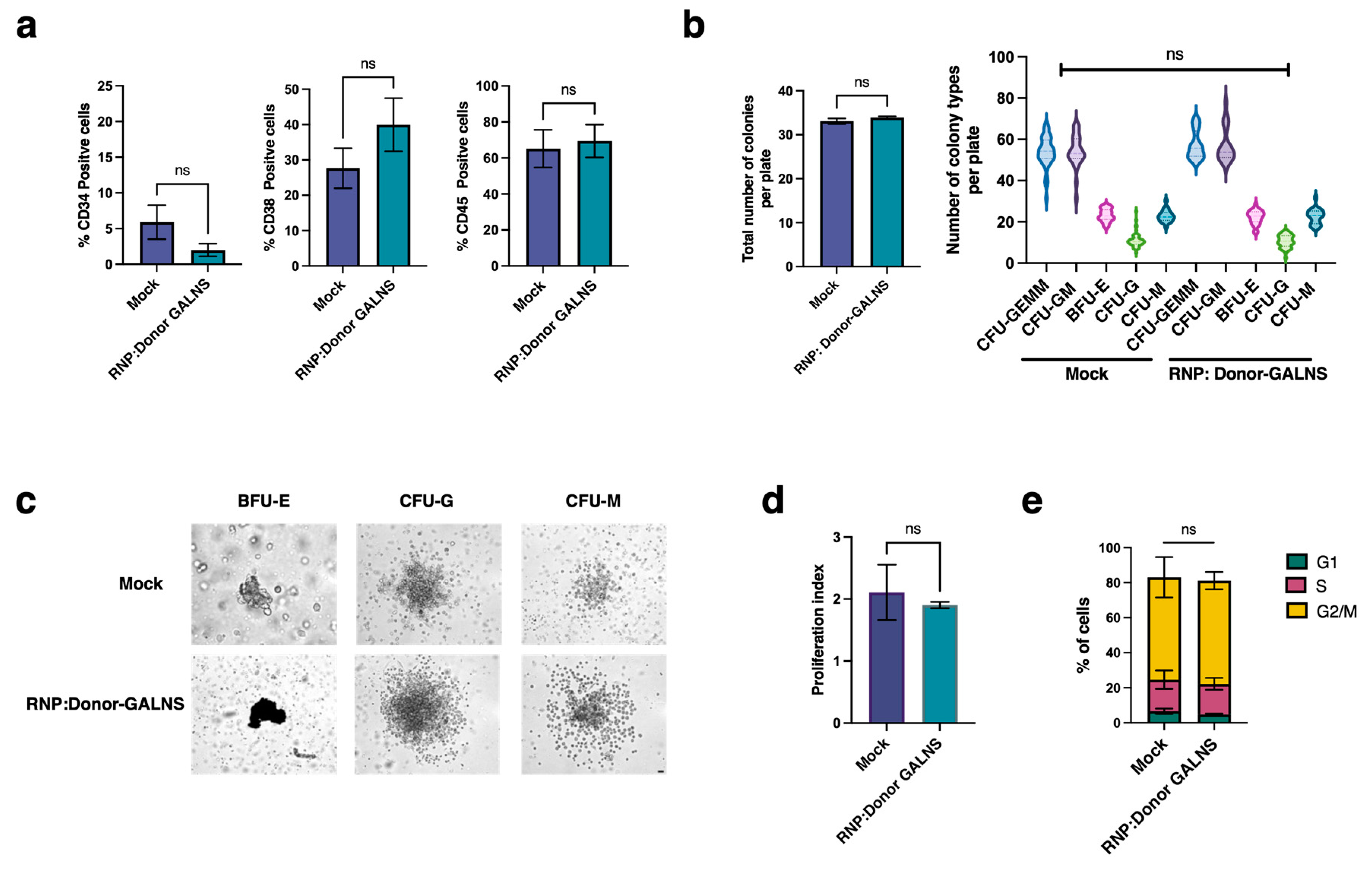
2.1. CRISPR/nCas9-Based Genome Editing System Efficiently Preserves the Self-Renewal and Differentiation Potential of CD34+ Cells
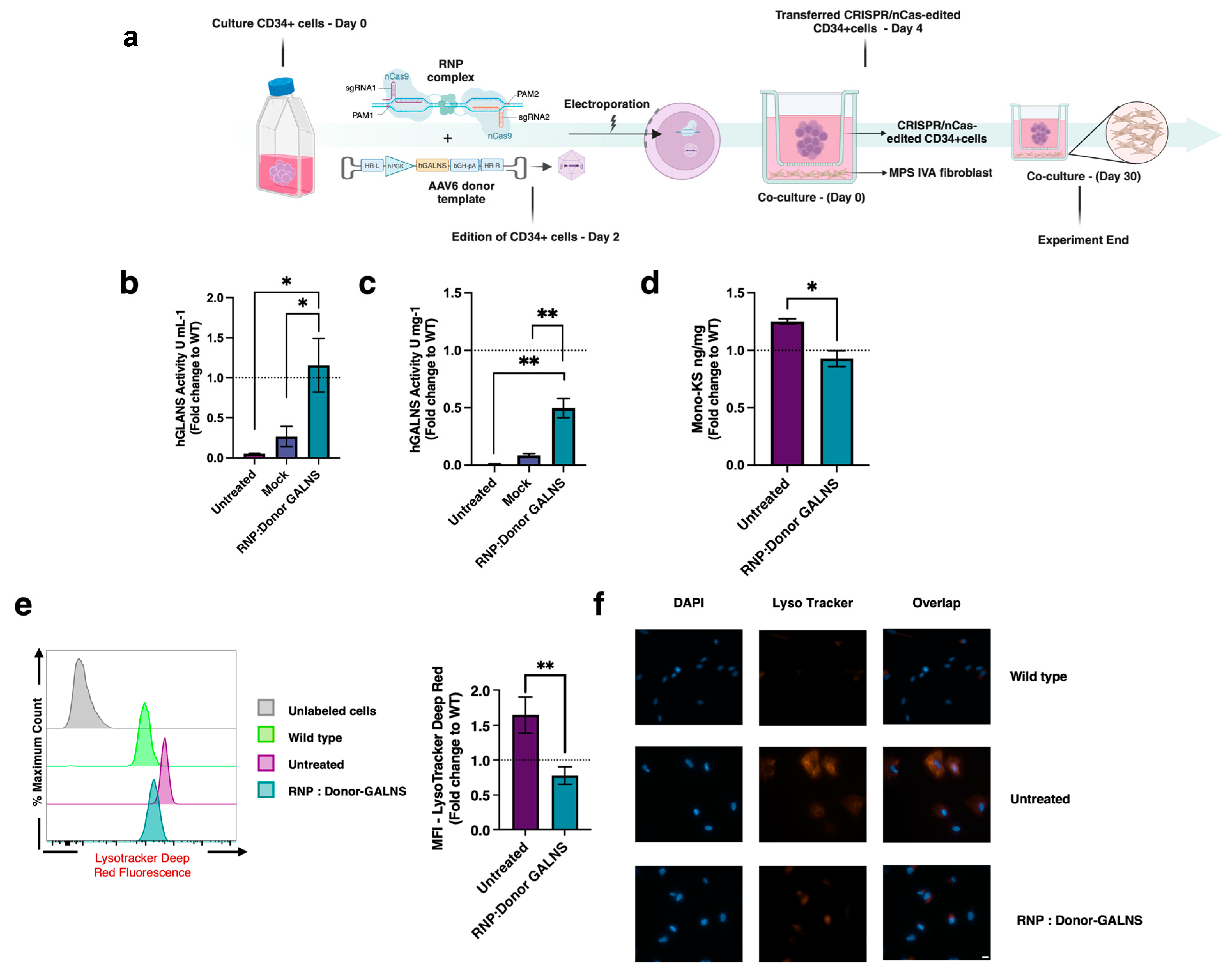
2.2. Therapeutic Efficacy of CRISPR/nCas9-Edited HSCs in MPS IVA Fibroblasts Co-Culture
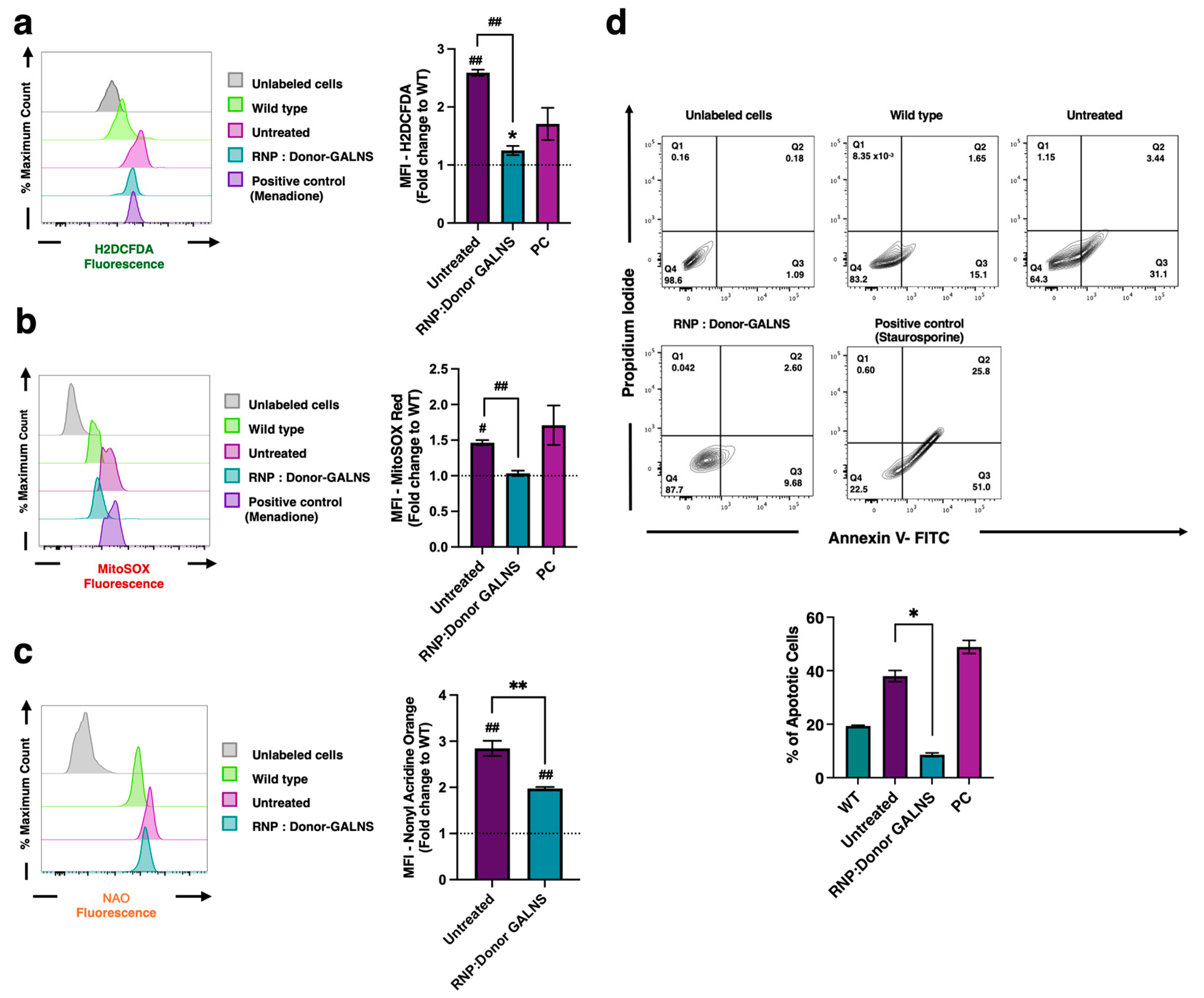
2.3. Reduction of Oxidative Stress in MPS IVA Fibroblasts
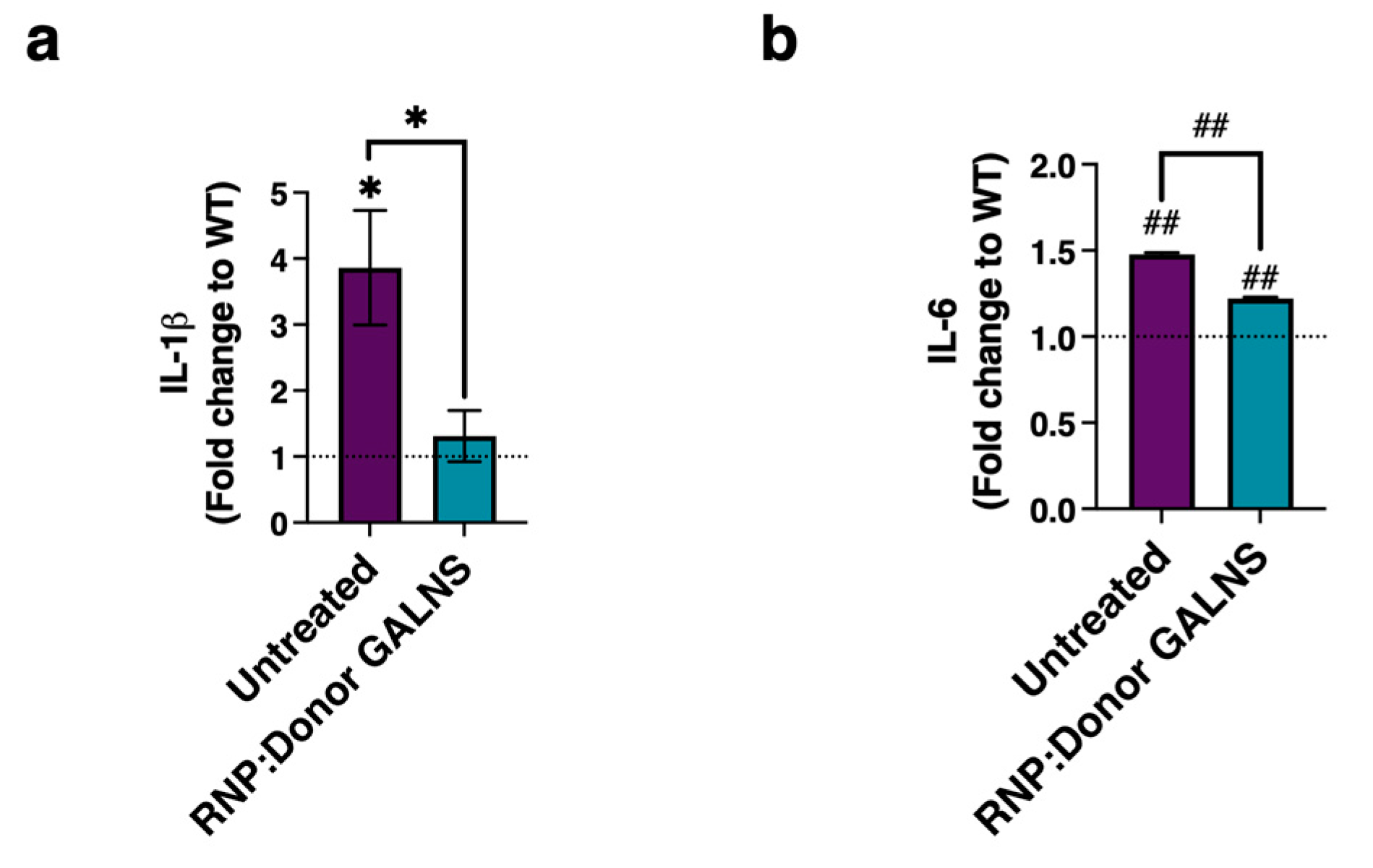
2.4. Decreased Levels of Pro-Inflammatory Markers in MPS IVA Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Expansion
4.2. Donor Template
4.3. CRISPR/nCas9 Editing Protocol
4.4. Donor Template Integration at the AAVS1 Locus
4.5. Stemness Evaluation
4.5.1. CD Markes
4.5.2. Colony-Forming Unit (CFU)
4.5.3. Proliferation and Cell Cycle Analysis
4.6. GALNS Activity Assays
4.7. Lysosomal Accumulation
4.7.1. Lysosomal Mass Evaluation
4.7.2. Analysis of Mono-Sulfated Keratan Sulfate
4.8. Oxidative Profile Assessment
4.8.1. Reactive Oxygen Species (ROS)
4.8.2. Mitochondrial-Derived Reactive Oxygen Species (mtROS)
4.9. Mitochondrial Mass Detection
4.10. Apoptosis Ratio
4.11. Pro-Inflammatory Profile Evaluation
5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| MPS | Mucopolysaccharidosis |
| LSD | Lysosomal storage disease |
| GAGs | Glycosaminoglycans |
| KS | Keratan sulfate |
| C6S | Chondroitin 6-sulfate |
| GALNS | N-acetylgalactosamine-6-sulfatase |
| GT | Gene therapy |
| ERT | Enzyme replacement therapy |
| HSC | Hematopoietic stem cell |
| HSCT | Hematopoietic stem cell transplantation |
| GVHD | Graft versus host disease |
| PC | Pharmacological chaperones |
| SDT | Substrate degradation therapy |
| WT | Wild type |
| EGFP | Enhanced green fluorescent protein |
References
- Sawamoto, K.; Álvarez González, J.V.; Piechnik, M.; Otero, F.J.; Couce, M.L.; Suzuki, Y.; Tomatsu, S. Mucopolysaccharidosis IVA: Diagnosis, Treatment, and Management. Int. J. Mol. Sci. 2020, 21, 1517. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Suzuki, Y.; Mackenzie, W.G.; Theroux, M.C.; Pizarro, C.; Yabe, H.; Orii, K.E.; Mason, R.W.; Orii, T.; Tomatsu, S. Current Therapies for Morquio a Syndrome and Their Clinical Outcomes. Expert Opin. Orphan Drugs 2016, 4, 941–951. [Google Scholar] [CrossRef]
- Khan, S.A.; Mason, R.W.; Giugliani, R.; Orii, K.; Fukao, T.; Suzuki, Y.; Yamaguchi, S.; Kobayashi, H.; Orii, T.; Tomatsu, S. Glycosaminoglycans Analysis in Blood and Urine of Patients with Mucopolysaccharidosis. Mol. Genet. Metab. 2018, 125, 44–52. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montano, A.M.; Oikawa, H.; Smith, M.; Barrera, L.; Chinen, Y.; Thacker, M.M.; Mackenzie, W.G.; Suzuki, Y.; Orii, T. Mucopolysaccharidosis Type Iva (Morquio a Disease): Clinical Review and Current Treatment. Curr. Pharm. Biotechnol. 2011, 12, 931–945. [Google Scholar] [CrossRef] [PubMed]
- Çelik, B.; Tomatsu, S.C.; Tomatsu, S.; Khan, S.A. Epidemiology of Mucopolysaccharidoses Update. Diagnostics 2021, 11, 273. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Almeciga-Diaz, C.J.; Tomatsu, S. Mucopolysaccharidosis IVA: Current Disease Models and Drawbacks. Int. J. Mol. Sci. 2023, 24, 6148. [Google Scholar] [CrossRef]
- Lee, C.-L.; Chuang, C.-K.; Chiu, H.-C.; Tu, R.-Y.; Lo, Y.-T.; Chang, Y.-H.; Lin, S.-P.; Lin, H.-Y. Clinical Utility of Elosulfase Alfa in the Treatment of Morquio A Syndrome. Drug Des. Dev. Ther. 2022, 16, 143–154. [Google Scholar] [CrossRef]
- Tomatsu, S.; Sawamoto, K.; Shimada, T.; Bober, M.B.; Kubaski, F.; Yasuda, E.; Mason, R.W.; Khan, S.; Alméciga-Díaz, C.J.; Barrera, L.A.; et al. Enzyme Replacement Therapy for Treating Mucopolysaccharidosis Type Iva (Morquio a Syndrome): Effect and Limitations. Expert Opin. Orphan Drugs 2015, 3, 1279–1290. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montano, A.M.; Ohashi, A.; Gutierrez, M.A.; Oikawa, H.; Oguma, T.; Dung, V.C.; Nishioka, T.; Orii, T.; Sly, W.S. Enzyme Replacement Therapy in a Murine Model of Morquio a Syndrome. Hum. Mol. Genet. 2008, 17, 815–824. [Google Scholar] [CrossRef]
- Tomatsu, S.; Montano, A.M.; Dung, V.C.; Ohashi, A.; Oikawa, H.; Oguma, T.; Orii, T.; Barrera, L.; Sly, W.S. Enhancement of Drug Delivery: Enzyme-Replacement Therapy for Murine Morquio a Syndrome. Mol. Ther. 2010, 18, 1094–1102. [Google Scholar] [CrossRef]
- Tomatsu, S.; Sawamoto, K.; Almeciga-Diaz, C.J.; Shimada, T.; Bober, M.B.; Chinen, Y.; Yabe, H.; Montano, A.M.; Giugliani, R.; Kubaski, F.; et al. Impact of Enzyme Replacement Therapy and Hematopoietic Stem Cell Transplantation in Patients with Morquio a Syndrome. Drug Des. Devel Ther. 2015, 9, 1937–1953. [Google Scholar] [CrossRef]
- Tomatsu, S.; Almeciga-Diaz, C.J.; Montano, A.M.; Yabe, H.; Tanaka, A.; Dung, V.C.; Giugliani, R.; Kubaski, F.; Mason, R.W.; Yasuda, E.; et al. Therapies for the Bone in Mucopolysaccharidoses. Mol. Genet. Metab. 2015, 114, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Do Cao, J.; Wiedemann, A.; Quinaux, T.; Battaglia-Hsu, S.F.; Mainard, L.; Froissart, R.; Bonnemains, C.; Ragot, S.; Leheup, B.; Journeau, P.; et al. 30 Months Follow-up of an Early Enzyme Replacement Therapy in a Severe Morquio a Patient: About One Case. Mol. Genet. Metab. Rep. 2016, 9, 42–45. [Google Scholar] [CrossRef] [PubMed]
- Doherty, C.; Stapleton, M.; Piechnik, M.; Mason, R.W.; Mackenzie, W.G.; Yamaguchi, S.; Kobayashi, H.; Suzuki, Y.; Tomatsu, S. Effect of Enzyme Replacement Therapy on the Growth of Patients with Morquio A. J. Hum. Genet. 2019, 64, 625–635. [Google Scholar] [CrossRef]
- Melbouci, M.; Mason, R.W.; Suzuki, Y.; Fukao, T.; Orii, T.; Tomatsu, S. Growth Impairment in Mucopolysaccharidoses. Mol. Genet. Metab. 2018, 124, 1–10. [Google Scholar] [CrossRef]
- Qi, Y.; Musson, D.G.; Schweighardt, B.; Tompkins, T.; Jesaitis, L.; Shaywitz, A.J.; Yang, K.; O’Neill, C.A. Pharmacokinetic and Pharmacodynamic Evaluation of Elosulfase Alfa, an Enzyme Replacement Therapy in Patients with Morquio a Syndrome. Clin. Pharmacokinet. 2014, 53, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Burrow, T.A.; Hopkin, R.J.; Leslie, N.D.; Tinkle, B.T.; Grabowski, G.A. Enzyme Reconstitution/Replacement Therapy for Lysosomal Storage Diseases. Curr. Opin. Pediatr. 2007, 19, 628–635. [Google Scholar] [CrossRef]
- Ponder, K.P. Immune Response Hinders Therapy for Lysosomal Storage Diseases. J. Clin. Investig. 2008, 118, 2686–2689. [Google Scholar] [CrossRef]
- Long, B.; Tompkins, T.; Decker, C.; Jesaitis, L.; Khan, S.; Slasor, P.; Harmatz, P.; O’Neill, C.A.; Schweighardt, B. Long-Term Immunogenicity of Elosulfase Alfa in the Treatment of Morquio a Syndrome: Results from Mor-005, a Phase III Extension Study. Clin. Ther. 2017, 39, 118–129.e3. [Google Scholar] [CrossRef]
- Schweighardt, B.; Tompkins, T.; Lau, K.; Jesaitis, L.; Qi, Y.; Musson, D.G.; Farmer, P.; Haller, C.; Shaywitz, A.J.; Yang, K.; et al. Immunogenicity of Elosulfase Alfa, an Enzyme Replacement Therapy in Patients with Morquio a Syndrome: Results from Mor-004, a Phase III Trial. Clin. Ther. 2015, 37, 1012–1021.e6. [Google Scholar] [CrossRef]
- Melton, A.C.; Soon, R.K., Jr.; Tompkins, T.; Long, B.; Schweighardt, B.; Qi, Y.; Vitelli, C.; Bagri, A.; Decker, C.; O’Neill, C.A.; et al. Antibodies That Neutralize Cellular Uptake of Elosulfase Alfa Are Not Associated with Reduced Efficacy or Pharmacodynamic Effect in Individuals with Morquio a Syndrome. J. Immunol. Methods 2017, 440, 41–51. [Google Scholar] [CrossRef]
- Cleary, M.; Davison, J.; Gould, R.; Geberhiwot, T.; Hughes, D.; Mercer, J.; Morrison, A.; Murphy, E.; Santra, S.; Jarrett, J.; et al. Impact of Long-Term Elosulfase Alfa Treatment on Clinical and Patient-Reported Outcomes in Patients with Mucopolysaccharidosis Type Iva: Results from a Managed Access Agreement in England. Orphanet J. Rare Dis. 2021, 16, 38. [Google Scholar] [CrossRef]
- Schlander, M.; Beck, M. Expensive Drugs for Rare Disorders: To Treat or Not to Treat? The Case of Enzyme Replacement Therapy for Mucopolysaccharidosis VI. Curr. Med. Res. Opin. 2009, 5, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Huang, Y.; Su, X.; Zhao, X.; Sheng, H.; Liang, C.; Jiang, M.; Zeng, C.; Cai, Y.; Lin, Y.; et al. Allogeneic Hematopoietic Stem Cell Transplantation for Mucopolysaccharidosis Patients: A Single-Center Experience and Assessment of Quality of Life. Ital. J. Pediatr. 2025, 51, 83. [Google Scholar] [CrossRef] [PubMed]
- Yabe, H.; Tanaka, A.; Chinen, Y.; Kato, S.; Sawamoto, K.; Yasuda, E.; Shintaku, H.; Suzuki, Y.; Orii, T.; Tomatsu, S. Hematopoietic Stem Cell Transplantation for Morquio a Syndrome. Mol. Genet. Metab. 2016, 117, 84–94. [Google Scholar] [CrossRef]
- Chinen, Y.; Higa, T.; Tomatsu, S.; Suzuki, Y.; Orii, T.; Hyakuna, N. Long-Term Therapeutic Efficacy of Allogenic Bone Marrow Transplantation in a Patient with Mucopolysaccharidosis Iva. Mol. Genet. Metab. Rep. 2014, 1, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Luan, Z.; Jiang, H.; Fang, J.; Qin, M.; Lee, V.; Chen, J. Allogeneic Hematopoietic Stem Cell Transplantation in Thirty-Four Pediatric Cases of Mucopolysaccharidosis-A Ten-Year Report from the China Children Transplant Group. Biol. Blood Marrow Transplant. 2016, 22, 2104–2108. [Google Scholar] [CrossRef]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Mason, R.W.; et al. Hematopoietic Stem Cell Transplantation for Mucopolysaccharidoses: Past, Present, and Future. Biol. Blood Marrow Transplant. J. Am. Soc. Blood Marrow Transplant. 2019, 25, e226–e246. [Google Scholar] [CrossRef]
- Yalcin, K.; Uygun, V.; Ozturk Hismi, B.; Celen, S.; Ozturkmen, S.; Zhumatayev, S.; Daloglu, H.; Karasu, G.; Yesilipek, A. Hematopoietic Stem Cell Transplantation in Children with Mucopolysaccharidosis Iva: Single Center Experience. Bone Marrow Transplant. 2025, 60, 47–51. [Google Scholar] [CrossRef]
- Xu, Z.L.; Huang, X.J. Optimizing Allogeneic Grafts in Hematopoietic Stem Cell Transplantation. Stem Cells Transl. Med. 2021, 10 (Suppl. S2), S41–S47. [Google Scholar] [CrossRef]
- Olarte-Avellaneda, S.; Cepeda Del Castillo, J.; Rojas-Rodriguez, A.F.; Sánchez, O.; Rodríguez-López, A.; Suárez García, D.A.; Pulido, L.M.S.; Alméciga-Díaz, C.J. Bromocriptine as a Novel Pharmacological Chaperone for Mucopolysaccharidosis Iva. ACS Med. Chem. Lett. 2020, 11, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Alméciga-Diaz, C.J.; Hidalgo, O.A.; Olarte-Avellaneda, S.; Rodríguez-López, A.; Guzman, E.; Garzón, R.; Pimentel-Vera, L.N.; Puentes-Tellez, M.A.; Rojas-Rodriguez, A.F.; Gorshkov, K.; et al. Identification of Ezetimibe and Pranlukast as Pharmacological Chaperones for the Treatment of the Rare Disease Mucopolysaccharidosis Type Iva. J. Med. Chem. 2019, 62, 6175–6189. [Google Scholar] [CrossRef]
- Sawamoto, K.; Tomatsu, S. Development of Substrate Degradation Enzyme Therapy for Mucopolysaccharidosis Iva Murine Model. Int. J. Mol. Sci. 2019, 20, 4139. [Google Scholar] [CrossRef]
- Rintz, E.; Celik, B.; Fnu, N.; Herreno-Pachon, A.M.; Khan, S.; Benincore-Florez, E.; Tomatsu, S. Molecular Therapy and Nucleic Acid Adeno-Associated Virus-Based Gene Therapy Delivering Combinations of Two Growth-Associated Genes to Mps Iva Mice. Mol. Ther. Nucleic Acids 2024, 35, 102211. [Google Scholar] [CrossRef] [PubMed]
- Celik, B.; Rintz, E.; Sansanwal, N.; Khan, S.; Bigger, B.; Tomatsu, S. Lentiviral Vector-Mediated ex Vivo hematopoietic Stem Cell Gene Therapy for Mucopolysaccharidosis Iva Murine Model. Hum. Gene Ther. 2024, 35, 917–937. [Google Scholar] [CrossRef] [PubMed]
- Leal, A.F.; Celik, B.; Fnu, N.; Khan, S.; Tomatsu, S.; Almeciga-Diaz, C.J. Iron Oxide-Coupled Crispr-Ncas9-Based Genome Editing Assessment in Mucopolysaccharidosis Iva Mice. Mol. Ther. Methods Clin. Dev. 2023, 31, 101153. [Google Scholar] [CrossRef]
- Leal, A.F.; Almeciga-Diaz, C.J. Efficient Crispr/Cas9 Nickase-Mediated Genome Editing in an In Vitro Model of Mucopolysaccharidosis Iva. Gene Ther. 2023, 30, 107–114. [Google Scholar] [CrossRef]
- Leal, A.F.; Cifuentes, J.; Torres, C.E.; Suarez, D.; Quezada, V.; Gomez, S.C.; Cruz, J.C.; Reyes, L.H.; Espejo-Mojica, A.J.; Almeciga-Diaz, C.J. Delivery and Assessment of a Crispr/Ncas9-Based Genome Editing System on In Vitro Models of Mucopolysaccharidoses Iva Assisted by Magnetite-Based Nanoparticles. Sci. Rep. 2022, 12, 15045. [Google Scholar] [CrossRef]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human Genome-Edited Hematopoietic Stem Cells Phenotypically Correct Mucopolysaccharidosis Type I. Nat. Commun. 2019, 10, 4045. [Google Scholar] [CrossRef]
- Scharenberg, S.G.; Poletto, E.; Lucot, K.L.; Colella, P.; Sheikali, A.; Montine, T.J.; Porteus, M.H.; Gomez-Ospina, N. Engineering Monocyte/Macrophage-Specific Glucocerebrosidase Expression in Human Hematopoietic Stem Cells Using Genome Editing. Nat. Commun. 2020, 11, 3327. [Google Scholar] [CrossRef]
- Cornetta, K.; Lin, T.Y.; Pellin, D.; Kohn, D.B. Meeting FDA Guidance Recommendations for Replication-Competent Virus and Insertional Oncogenesis Testing. Mol. Ther. Methods Clin. Dev. 2023, 28, 28–39. [Google Scholar] [CrossRef]
- Schlimgen, R.; Howard, J.; Wooley, D.; Thompson, M.; Baden, L.R.; Yang, O.O.; Christiani, D.C.; Mostoslavsky, G.; Diamond, D.V.; Duane, E.G.; et al. Risks Associated with Lentiviral Vector Exposures and Prevention Strategies. J. Occup. Environ. Med. 2016, 58, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.; Ott, M.G.; Schultze-Strasser, S.; Jauch, A.; Burwinkel, B.; Kinner, A.; Schmidt, M.; Krämer, A.; Schwäble, J.; Glimm, H.; et al. Genomic Instability and Myelodysplasia with Monosomy 7 Consequent to Evi1 Activation after Gene Therapy for Chronic Granulomatous Disease. Nat. Med. 2010, 16, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Duncan, C.N.; Bledsoe, J.R.; Grzywacz, B.; Beckman, A.; Bonner, M.; Eichler, F.S.; Kühl, J.S.; Harris, M.H.; Slauson, S.; Colvin, R.A.; et al. Hematologic Cancer after Gene Therapy for Cerebral Adrenoleukodystrophy. N. Engl. J. Med. 2024, 391, 1287–1301. [Google Scholar] [CrossRef] [PubMed]
- Riesenberg, S.; Kanis, P.; Macak, D.; Wollny, D.; Düsterhöft, D.; Kowalewski, J.; Helmbrecht, N.; Maricic, T.; Pääbo, S. Efficient High-Precision Homology-Directed Repair-Dependent Genome Editing by Hdrobust. Nat. Methods 2023, 20, 1388–1399. [Google Scholar] [CrossRef]
- Poletto, E.; Baldo, G.; Gomez-Ospina, N. Genome Editing for Mucopolysaccharidoses. Int. J. Mol. Sci. 2020, 21, 500. [Google Scholar] [CrossRef]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A High-Fidelity Cas9 Mutant Delivered as a Ribonucleoprotein Complex Enables Efficient Gene Editing in Human Hematopoietic Stem and Progenitor Cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. Crispr-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef]
- Chiang, T.W.; le Sage, C.; Larrieu, D.; Demir, M.; Jackson, S.P. Crispr-Cas9(D10a) Nickase-Based Genotypic and Phenotypic Screening to Enhance Genome Editing. Sci. Rep. 2016, 6, 24356. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double Nicking by Rna-Guided Crispr Cas9 for Enhanced Genome Editing Specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef]
- Leal, A.F.; Benincore-Florez, E.; Rintz, E.; Herreno-Pachon, A.M.; Celik, B.; Ago, Y.; Almeciga-Diaz, C.J.; Tomatsu, S. Mucopolysaccharidoses: Cellular Consequences of Glycosaminoglycans Accumulation and Potential Targets. Int. J. Mol. Sci. 2022, 24, 477. [Google Scholar] [CrossRef] [PubMed]
- Fecarotta, S.; Tarallo, A.; Damiano, C.; Minopoli, N.; Parenti, G. Pathogenesis of Mucopolysaccharidoses, an Update. Int. J. Mol. Sci. 2020, 21, 2515. [Google Scholar] [CrossRef] [PubMed]
- Gaffke, L.; Szczudlo, Z.; Podlacha, M.; Cyske, Z.; Rintz, E.; Mantej, J.; Krzelowska, K.; Wegrzyn, G.; Pierzynowska, K. Impaired ion Homeostasis as a Possible Associate Factor in Mucopolysaccharidosis Pathogenesis: Transcriptomic, Cellular and Animal Studies. Metab. Brain Dis. 2022, 37, 299–310. [Google Scholar] [CrossRef]
- Leal, A.F.; Cifuentes, J.; Quezada, V.; Benincore-Florez, E.; Cruz, J.C.; Reyes, L.H.; Espejo-Mojica, A.J.; Almeciga-Diaz, C.J. Crispr/Ncas9-Based Genome Editing on Gm2 Gangliosidoses Fibroblasts via Non-Viral Vectors. Int. J. Mol. Sci. 2022, 23, 672. [Google Scholar] [CrossRef]
- Loor, G.; Kondapalli, J.; Schriewer, J.M.; Chandel, N.S.; Vanden Hoek, T.L.; Schumacker, P.T. Menadione Triggers Cell Death through Ros-Dependent Mechanisms Involving Parp Activation without Requiring Apoptosis. Free Radic. Biol. Med. 2010, 49, 1925–1936. [Google Scholar] [CrossRef]
- Puentes-Tellez, M.A.; Sanchez, O.F.; Rojas-Rodriguez, F.; Benincore-Florez, E.; Barbosa, H.; Almeciga Diaz, C.J. Evaluation of Hiv-1 Derived Lentiviral Vectors as Transductors of Mucopolysaccharidosis Type Iv a Fibroblasts. Gene 2021, 780, 145527. [Google Scholar] [CrossRef]
- Kuk, M.U.; Lee, Y.H.; Kim, J.W.; Hwang, S.Y.; Park, J.T.; Park, S.C. Potential Treatment of Lysosomal Storage Disease through Modulation of the Mitochondrial-Lysosomal Axis. Cells 2021, 10, 420. [Google Scholar] [CrossRef] [PubMed]
- Stepien, K.M.; Roncaroli, F.; Turton, N.; Hendriksz, C.J.; Roberts, M.; Heaton, R.A.; Hargreaves, I. Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. J. Clin. Med. 2020, 9, 2596. [Google Scholar] [CrossRef]
- Brokowska, J.; Gaffke, L.; Pierzynowska, K.; Wegrzyn, G. Enhanced Efficiency of the Basal and Induced Apoptosis Process in Mucopolysaccharidosis Iva and Ivb Human Fibroblasts. Int. J. Mol. Sci. 2023, 24, 4119. [Google Scholar] [CrossRef]
- Antonsson, A.; Persson, J.L. Induction of Apoptosis by Staurosporine Involves the Inhibition of Expression of the Major Cell Cycle Proteins at the G(2)/M Checkpoint Accompanied by Alterations in Erk and Akt Kinase Activities. Anticancer Res. 2009, 29, 2893–2898. [Google Scholar]
- Fujitsuka, H.; Sawamoto, K.; Peracha, H.; Mason, R.W.; Mackenzie, W.; Kobayashi, H.; Yamaguchi, S.; Suzuki, Y.; Orii, K.; Orii, T.; et al. Biomarkers in Patients with Mucopolysaccharidosis Type Ii and Iv. Mol. Genet. Metab. Rep. 2019, 19, 100455. [Google Scholar] [CrossRef] [PubMed]
- Davison, J. How to Use…White Blood Cell Enzyme Assays. Arch. Dis. Child. Educ. Pract. Ed. 2017, 102, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Donida, B.; Marchetti, D.P.; Biancini, G.B.; Deon, M.; Manini, P.R.; da Rosa, H.T.; Moura, D.J.; Saffi, J.; Bender, F.; Burin, M.G.; et al. Oxidative Stress and Inflammation in Mucopolysaccharidosis Type Iva Patients Treated with Enzyme Replacement Therapy. Biochim. Biophys. Acta 2015, 1852, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Ago, Y.; Rintz, E.; Musini, K.S.; Ma, Z.; Tomatsu, S. Molecular Mechanisms in Pathophysiology of Mucopolysaccharidosis and Prospects for Innovative Therapy. Int. J. Mol. Sci. 2024, 25, 1113. [Google Scholar] [CrossRef]
- Yamamoto, R.; Wilkinson, A.C.; Nakauchi, H. In Vivo and Ex Vivo Haematopoietic Stem Cell Expansion. Curr. Opin. Hematol. 2020, 27, 273–278. [Google Scholar] [CrossRef]
- Koniali, L.; Lederer, C.W.; Kleanthous, M. Therapy Development by Genome Editing of Hematopoietic Stem Cells. Cells 2021, 10, 1492. [Google Scholar] [CrossRef]
- Jeong, Y.S.; Kim, E.J.; Shim, C.K.; Hou, J.H.; Kim, J.M.; Choi, H.G.; Kim, W.K.; Oh, Y.K. Modulation of Biodistribution and Expression of Plasmid DNA Following Mesenchymal Progenitor Cell-Based Delivery. J. Drug Target. 2008, 16, 405–414. [Google Scholar] [CrossRef]
- Wang, Z.; Troilo, P.J.; Wang, X.; Griffiths, T.G.; Pacchione, S.J.; Barnum, A.B.; Harper, L.B.; Pauley, C.J.; Niu, Z.; Denisova, L.; et al. Detection of Integration of Plasmid DNA into Host Genomic DNA Following Intramuscular Injection and Electroporation. Gene Ther. 2004, 11, 711–721. [Google Scholar] [CrossRef]
- Maita, N.; Tsukimura, T.; Taniguchi, T.; Saito, S.; Ohno, K.; Taniguchi, H.; Sakuraba, H. Human α-L-Iduronidase Uses Its Own N-Glycan as a Substrate-Binding and Catalytic Module. Proc. Natl. Acad. Sci. USA 2013, 110, 14628–14633. [Google Scholar] [CrossRef]
- Bie, H.; Yin, J.; He, X.; Kermode, A.R.; Goddard-Borger, E.D.; Withers, S.G.; James, M.N. Insights into Mucopolysaccharidosis I from the Structure and Action of Alpha-L-Iduronidase. Nat. Chem. Biol. 2013, 9, 739–745. [Google Scholar] [CrossRef]
- Rivera-Colon, Y.; Schutsky, E.K.; Kita, A.Z.; Garman, S.C. The Structure of Human Galns Reveals the Molecular Basis for Mucopolysaccharidosis Iv A. J. Mol. Biol. 2012, 423, 736–751. [Google Scholar] [CrossRef] [PubMed]
- van Rensburg, R.; Beyer, I.; Yao, X.Y.; Wang, H.; Denisenko, O.; Li, Z.Y.; Russell, D.W.; Miller, D.G.; Gregory, P.; Holmes, M.; et al. Chromatin Structure of Two Genomic Sites for Targeted Transgene Integration in Induced Pluripotent Stem Cells and Hematopoietic Stem Cells. Gene Ther. 2013, 20, 201–214. [Google Scholar] [CrossRef]
- Wagenblast, E.; Azkanaz, M.; Smith, S.A.; Shakib, L.; McLeod, J.L.; Krivdova, G.; Araujo, J.; Shultz, L.D.; Gan, O.I.; Dick, J.E.; et al. Functional Profiling of Single Crispr/Cas9-Edited Human Long-Term Hematopoietic Stem Cells. Nat. Commun. 2019, 10, 4730. [Google Scholar] [CrossRef]
- Bak, R.O.; Dever, D.P.; Porteus, M.H. Crispr/Cas9 Genome Editing in Human Hematopoietic Stem Cells. Nat. Protoc. 2018, 13, 358–376. [Google Scholar] [CrossRef]
- Bak, R.O.; Dever, D.P.; Reinisch, A.; Cruz Hernandez, D.; Majeti, R.; Porteus, M.H. Multiplexed Genetic Engineering of Human Hematopoietic Stem and Progenitor Cells Using CRISPR/Cas9 and AAV6. eLife 2017, 6, e27873. [Google Scholar] [CrossRef] [PubMed]
- Bloomer, H.; Smith, R.H.; Hakami, W.; Larochelle, A. Genome Editing in Human Hematopoietic Stem and Progenitor Cells Via Crispr-Cas9-Mediated Homology-Independent Targeted Integration. Mol. Ther. 2021, 29, 1611–1624. [Google Scholar] [CrossRef]
- Rai, R.; Naseem, A.; Vetharoy, W.; Steinberg, Z.; Thrasher, A.J.; Santilli, G.; Cavazza, A. An Improved Medium Formulation for Efficient Ex Vivo Gene Editing, Expansion and Engraftment of Hematopoietic Stem and Progenitor Cells. Mol. Ther. Methods Clin. Dev. 2023, 29, 58–69. [Google Scholar] [CrossRef]
- Cannon, P.; Asokan, A.; Czechowicz, A.; Hammond, P.; Kohn, D.B.; Lieber, A.; Malik, P.; Marks, P.; Porteus, M.; Verhoeyen, E.; et al. Safe and Effective in Vivo Targeting and Gene Editing in Hematopoietic Stem Cells: Strategies for Accelerating Development. Hum. Gene Ther. 2021, 32, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Consiglieri, G.; Bernardo, M.E.; Brunetti-Pierri, N.; Aiuti, A. Ex vivo and In vivo Gene Therapy for Mucopolysaccharidoses: State of the Art. Hematol. Oncol. Clin. North Am. 2022, 36, 865–878. [Google Scholar] [CrossRef]
- Pierzynowska, K.; Gaffke, L.; Cyske, Z.; Wegrzyn, G.; Buttari, B.; Profumo, E.; Saso, L. Oxidative Stress in Mucopolysaccharidoses: Pharmacological Implications. Molecules 2021, 26, 5616. [Google Scholar] [CrossRef]
- Oliveira-Silva, J.A.; Yamamoto, J.U.P.; Oliveira, R.B.; Monteiro, V.C.L.; Frangipani, B.J.; Kyosen, S.O.; Martins, A.M.; D’Almeida, V. Oxidative Stress Assessment by Glutathione Peroxidase Activity and Glutathione Levels in Response to Selenium Supplementation in Patients with Mucopolysaccharidosis I, Ii and Vi. Genet. Mol. Biol. 2019, 42, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (Ros) and Ros-Induced Ros Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial Dysfunction and Oxidative Stress in Metabolic Disorders—A Step Towards Mitochondria Based Therapeutic Strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Tillo, M.; Lamanna, W.C.; Dwyer, C.A.; Sandoval, D.R.; Pessentheiner, A.R.; Al-Azzam, N.; Sarrazin, S.; Gonzales, J.C.; Kan, S.H.; Andreyev, A.Y.; et al. Impaired Mitophagy in Sanfilippo a Mice Causes Hypertriglyceridemia and Brown Adipose Tissue Activation. J. Biol. Chem. 2022, 298, 102159. [Google Scholar] [CrossRef]
- Liu, D.; Cai, Z.J.; Yang, Y.T.; Lu, W.H.; Pan, L.Y.; Xiao, W.F.; Li, Y.S. Mitochondrial Quality Control in Cartilage Damage and Osteoarthritis: New Insights and Potential Therapeutic Targets. Osteoarthr. Cartil. 2022, 30, 395–405. [Google Scholar] [CrossRef]
- Obeng, E. Apoptosis (Programmed Cell Death) and Its Signals—A Review. Braz. J. Biol. 2021, 81, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Donida, B.; Marchetti, D.P.; Jacques, C.E.D.; Ribas, G.; Deon, M.; Manini, P.; da Rosa, H.T.; Moura, D.J.; Saffi, J.; Giugliani, R.; et al. Oxidative Profile Exhibited by Mucopolysaccharidosis Type Iva Patients at Diagnosis: Increased Keratan Urinary Levels. Mol. Genet. Metab. Rep. 2017, 11, 46–53. [Google Scholar] [CrossRef]
- Wiesinger, A.M.; Bigger, B.; Giugliani, R.; Scarpa, M.; Moser, T.; Lampe, C.; Kampmann, C.; Lagler, F.B. The Inflammation in the Cytopathology of Patients with Mucopolysaccharidoses—Immunomodulatory Drugs as an Approach to Therapy. Front. Pharmacol. 2022, 13, 863667. [Google Scholar] [CrossRef]
- Bozhilov, Y.K.; Hsu, I.; Brown, E.J.; Wilkinson, A.C. In Vitro Human Haematopoietic Stem Cell Expansion and Differentiation. Cells 2023, 12, 896. [Google Scholar] [CrossRef]
- Vignon, C.; Debeissat, C.; Georget, M.T.; Bouscary, D.; Gyan, E.; Rosset, P.; Herault, O. Flow Cytometric Quantification of All Phases of the Cell Cycle and Apoptosis in a Two-Color Fluorescence Plot. PLoS ONE 2013, 8, e68425. [Google Scholar] [CrossRef]
- Toietta, G.; Severini, G.M.; Traversari, C.; Tomatsu, S.; Sukegawa, K.; Fukuda, S.; Kondo, N.; Tortora, P.; Bordignon, C. Various Cells Retrovirally Transduced with N-Acetylgalactosoamine-6-Sulfate Sulfatase Correct Morquio Skin Fibroblasts in Vitro. Hum. Gene Ther. 2001, 12, 2007–2016. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Sawamoto, K.; Karumuthil-Melethil, S.; Khan, S.; Stapleton, M.; Bruder, J.T.; Danos, O.; Tomatsu, S. Liver-Targeted Aav8 Gene Therapy Ameliorates Skeletal and Cardiovascular Pathology in a Mucopolysaccharidosis Iva Murine Model. Mol. Ther. Methods Clin. Dev. 2020, 18, 50–61. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herreno-Pachón, A.M.; Leal, A.F.; Khan, S.; Alméciga-Díaz, C.J.; Tomatsu, S. CRISPR/nCas9-Edited CD34+ Cells Rescue Mucopolysaccharidosis IVA Fibroblasts Phenotype. Int. J. Mol. Sci. 2025, 26, 4334. https://doi.org/10.3390/ijms26094334
Herreno-Pachón AM, Leal AF, Khan S, Alméciga-Díaz CJ, Tomatsu S. CRISPR/nCas9-Edited CD34+ Cells Rescue Mucopolysaccharidosis IVA Fibroblasts Phenotype. International Journal of Molecular Sciences. 2025; 26(9):4334. https://doi.org/10.3390/ijms26094334
Chicago/Turabian StyleHerreno-Pachón, Angélica María, Andrés Felipe Leal, Shaukat Khan, Carlos Javier Alméciga-Díaz, and Shunji Tomatsu. 2025. "CRISPR/nCas9-Edited CD34+ Cells Rescue Mucopolysaccharidosis IVA Fibroblasts Phenotype" International Journal of Molecular Sciences 26, no. 9: 4334. https://doi.org/10.3390/ijms26094334
APA StyleHerreno-Pachón, A. M., Leal, A. F., Khan, S., Alméciga-Díaz, C. J., & Tomatsu, S. (2025). CRISPR/nCas9-Edited CD34+ Cells Rescue Mucopolysaccharidosis IVA Fibroblasts Phenotype. International Journal of Molecular Sciences, 26(9), 4334. https://doi.org/10.3390/ijms26094334