
1,3-Dipolar Cycloaddition and Mannich Reactions of Alkynyl Triterpenes: New Trends in Synthetic Strategies and Pharmacological Applications

Abstract
1. Introduction
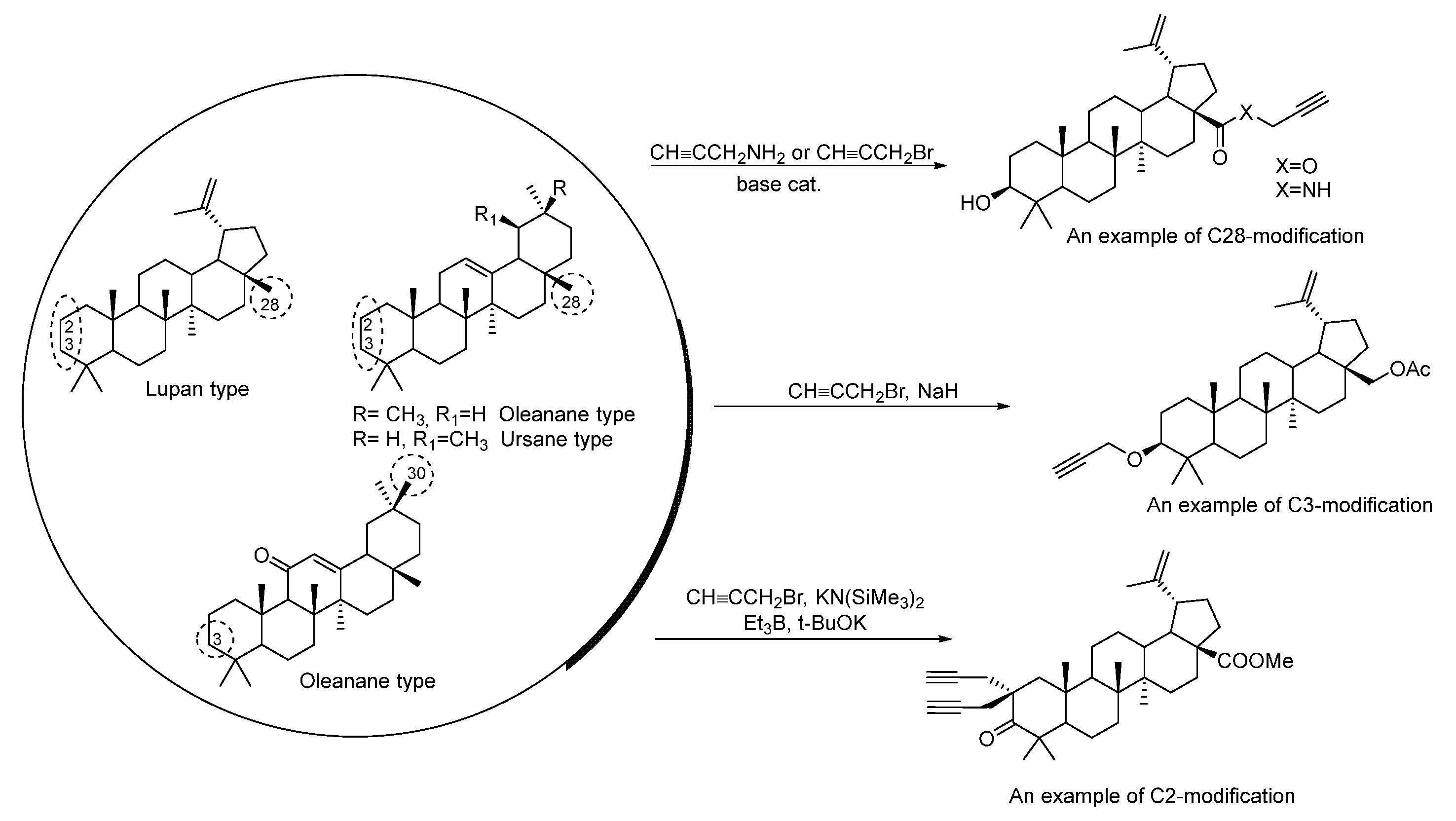
2. Synthesis of Triterpene Alkynes and Azides as the Key Precursors for 1,2,3-Triazoles and Mannich Bases
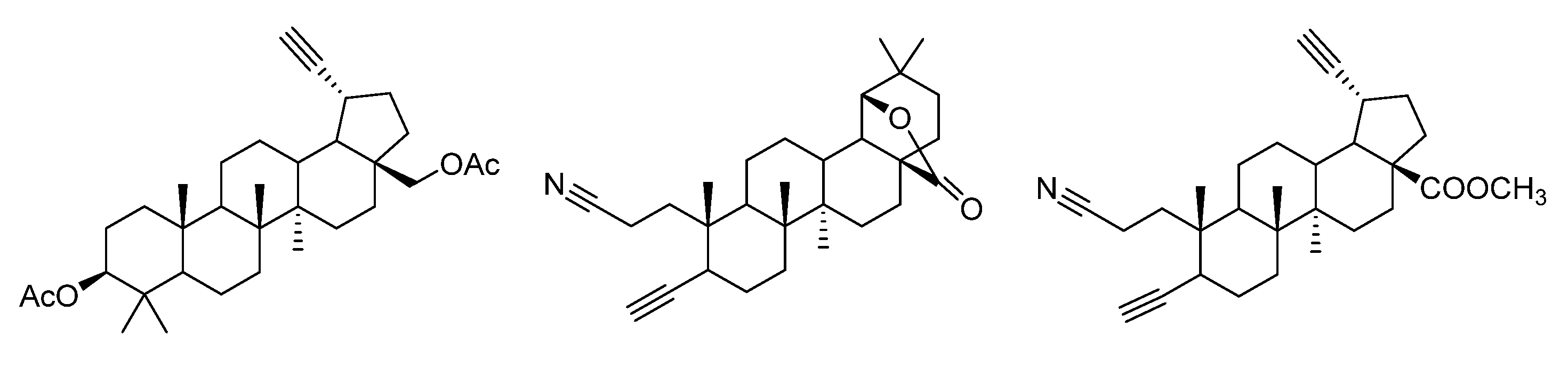
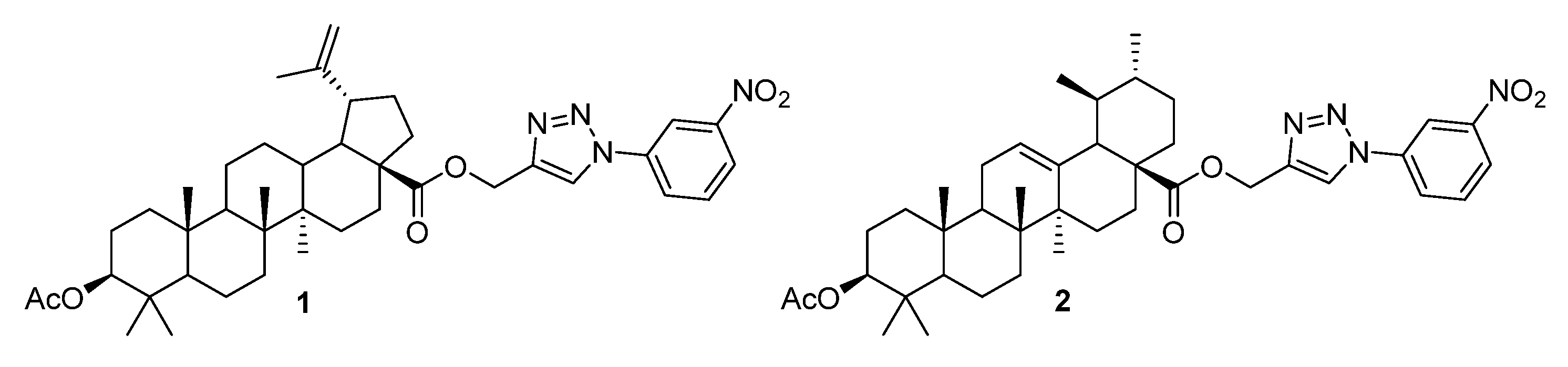
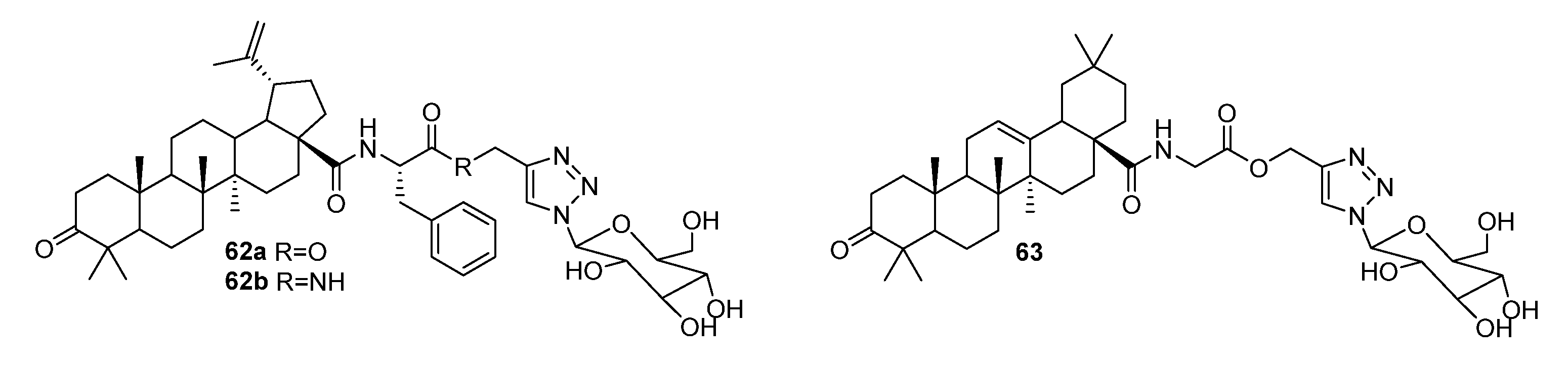
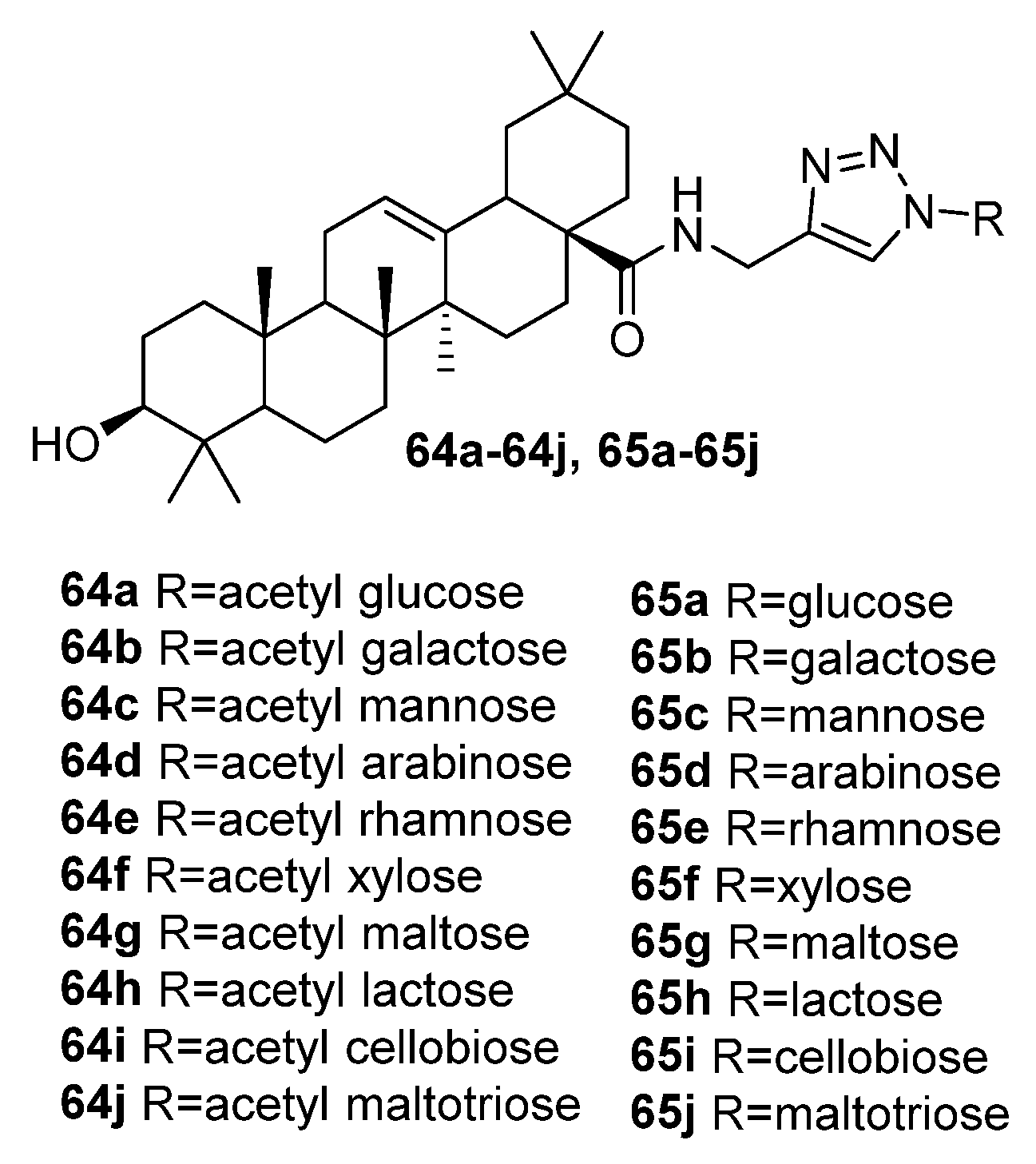
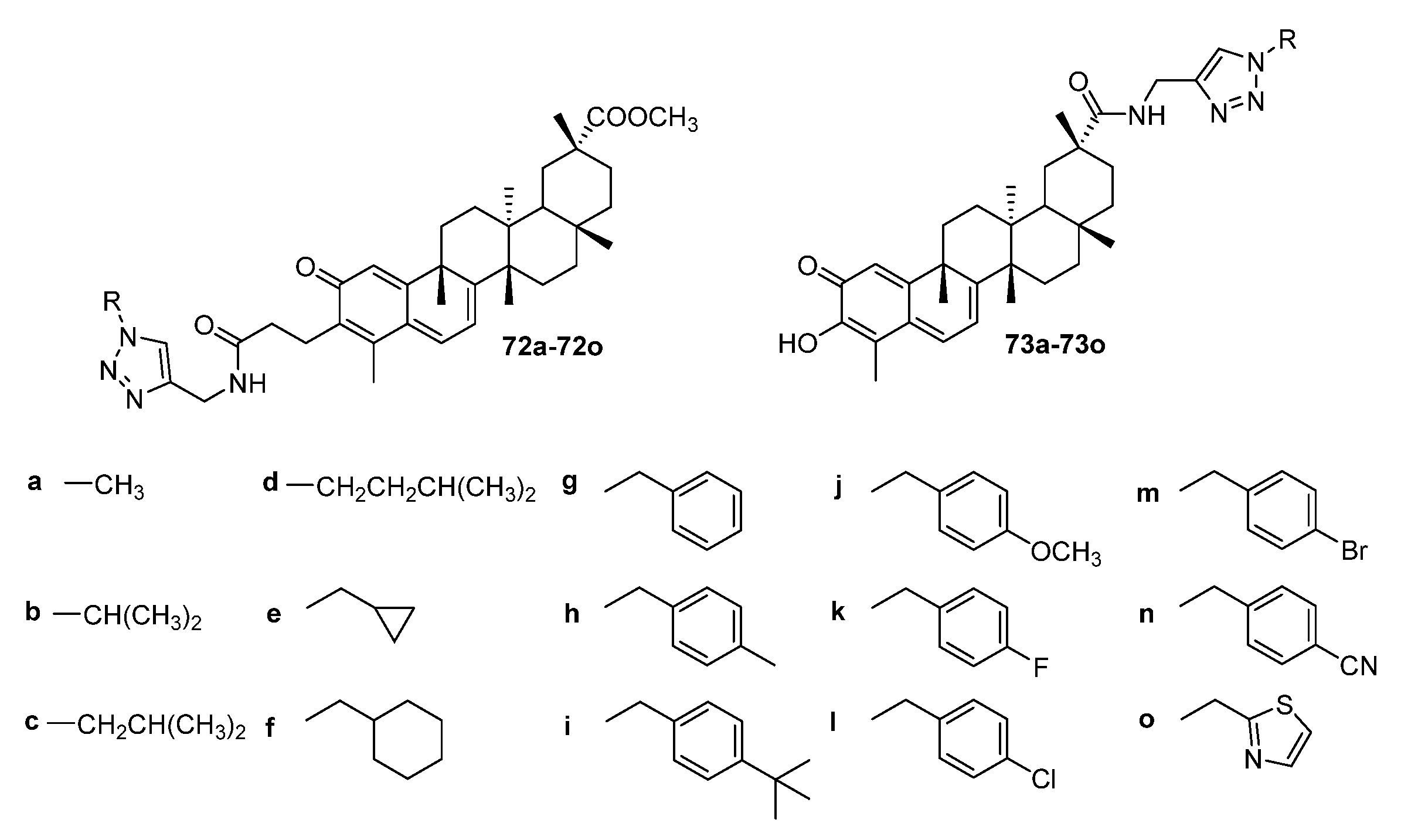
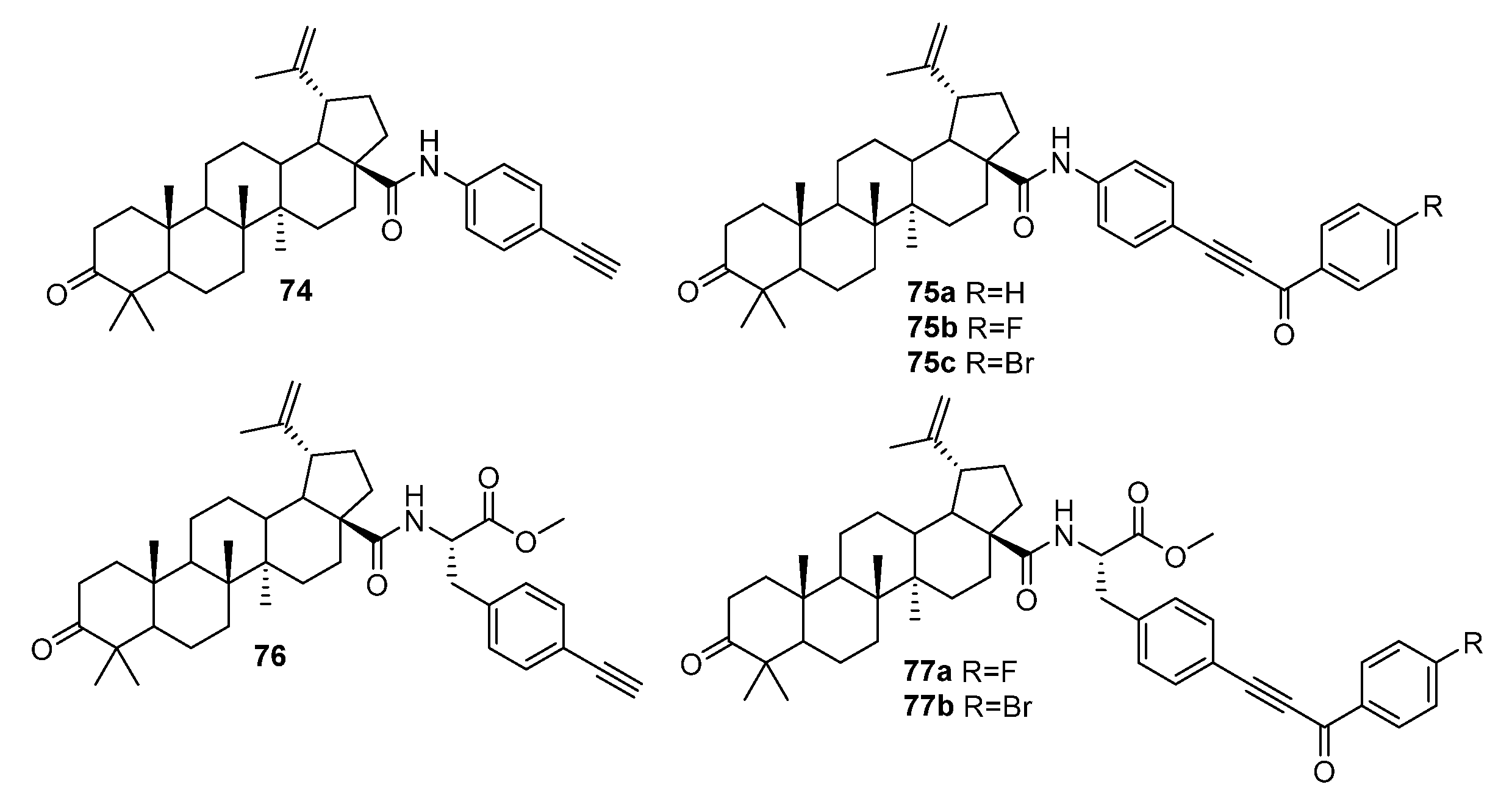
3. Modification of Alkynyl-Triterpenoids by the Click Reaction
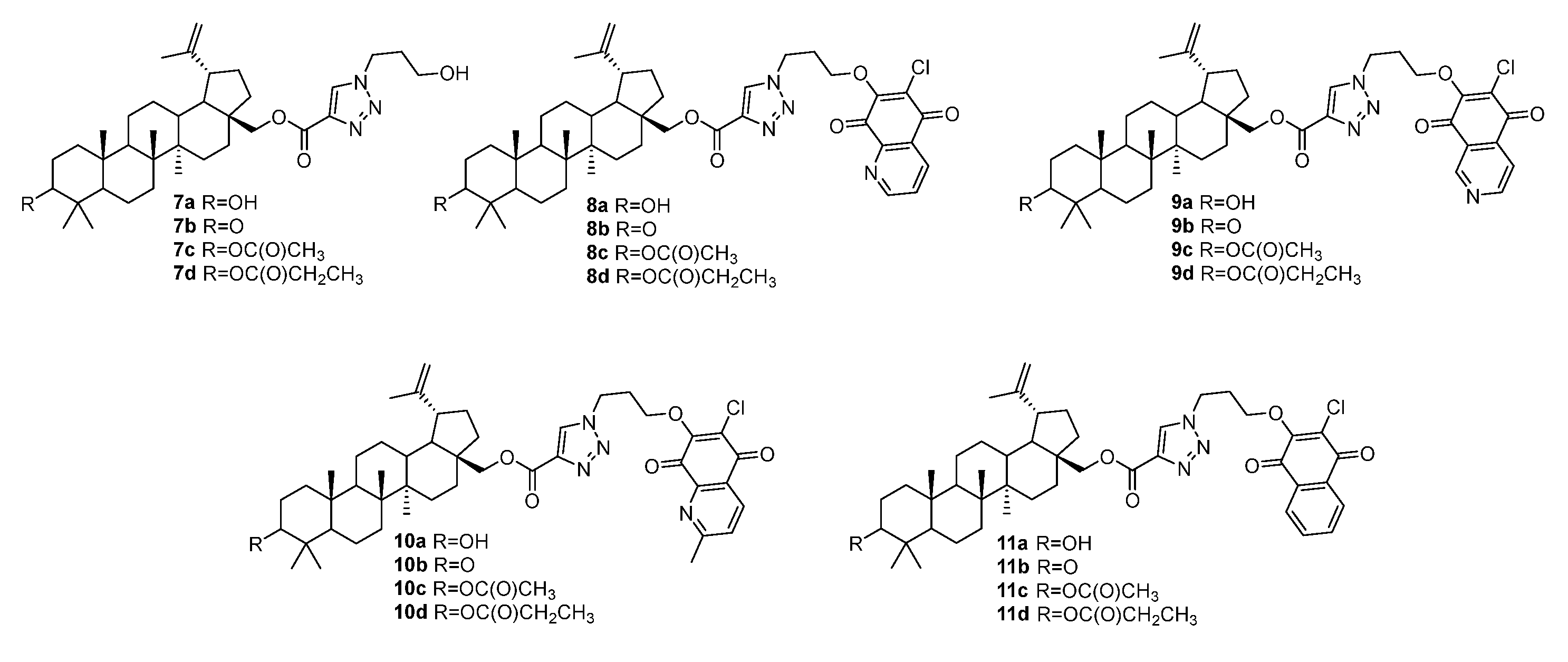
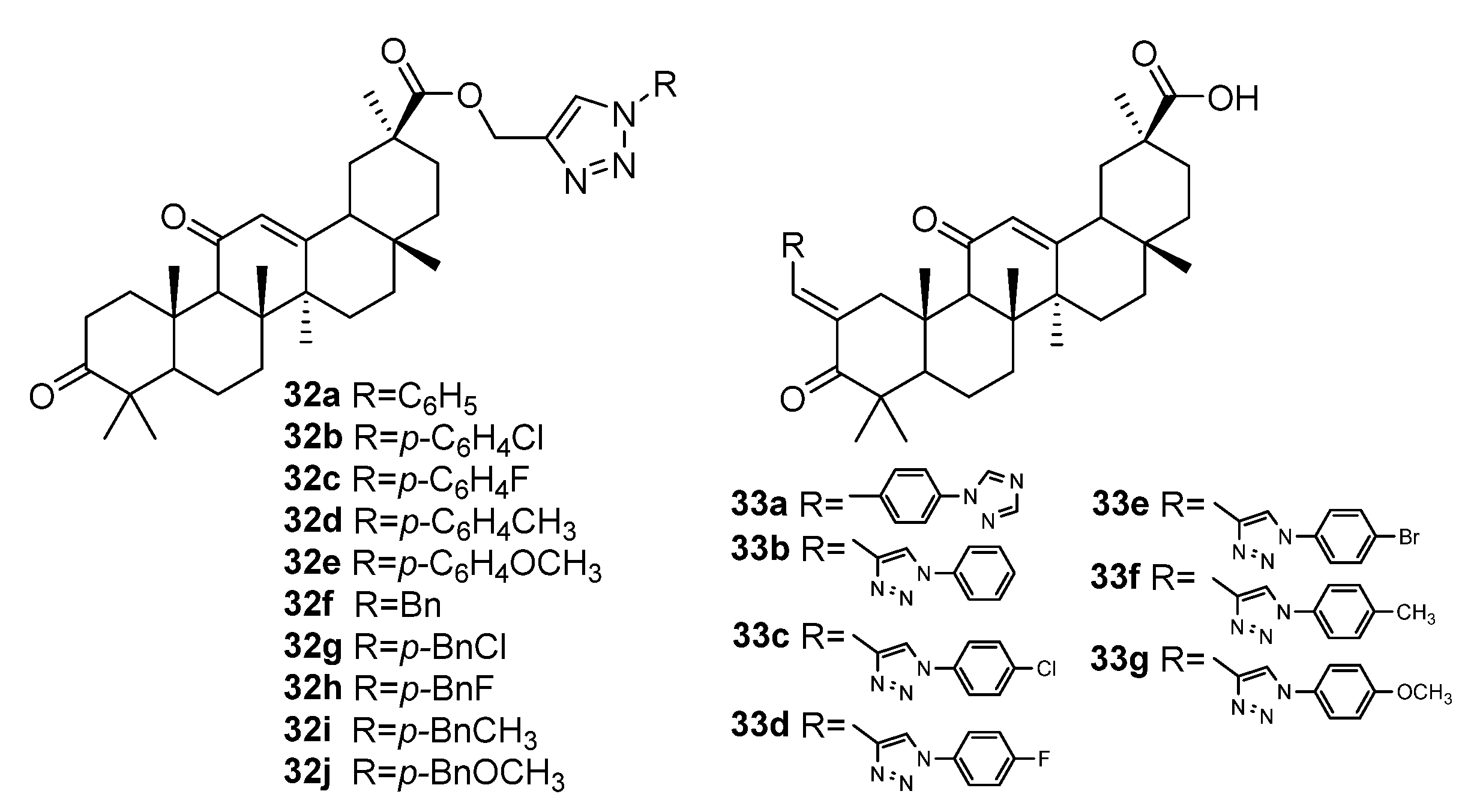
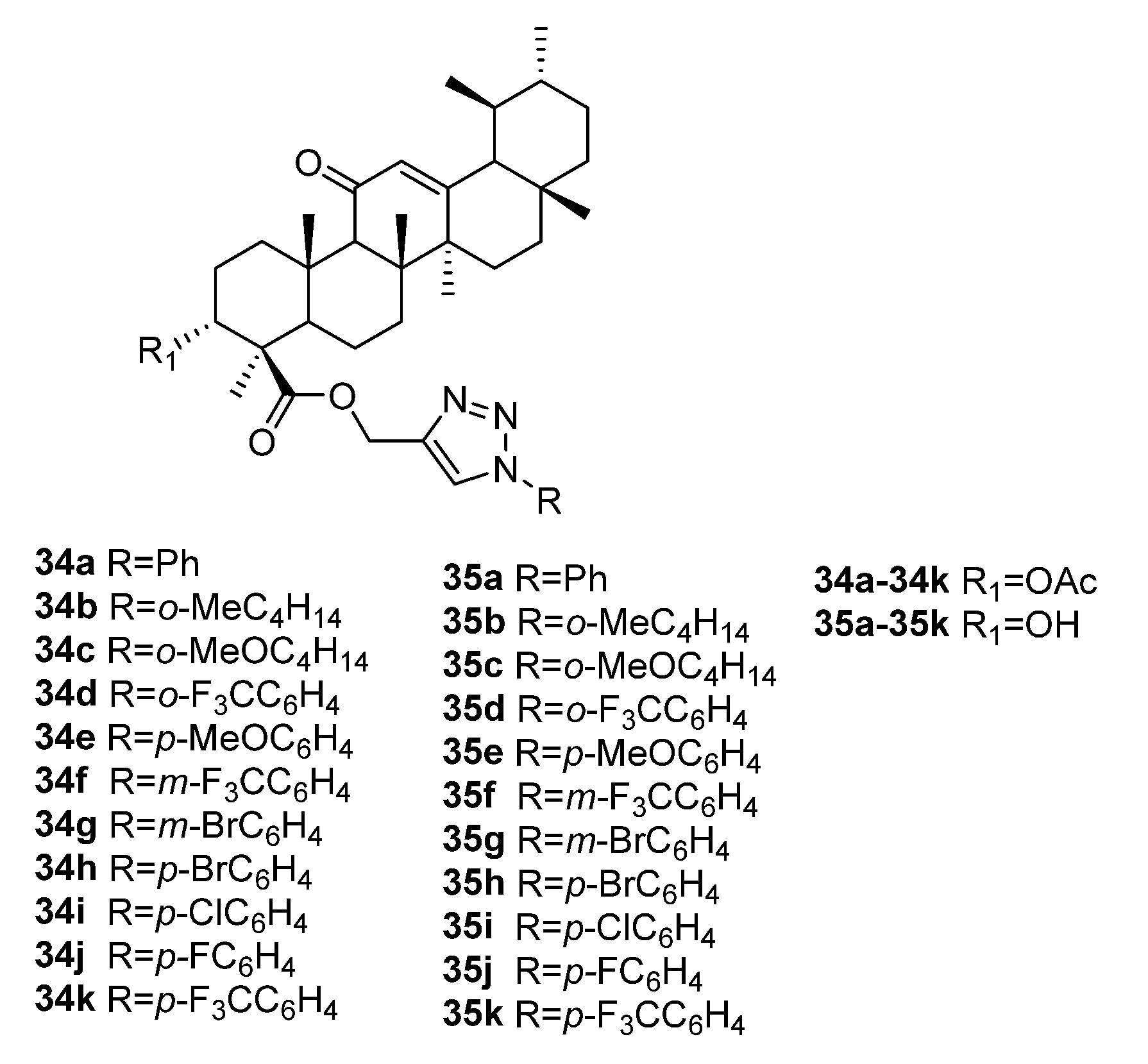
3.1. Modifications at C28/C30 Carboxylic Position
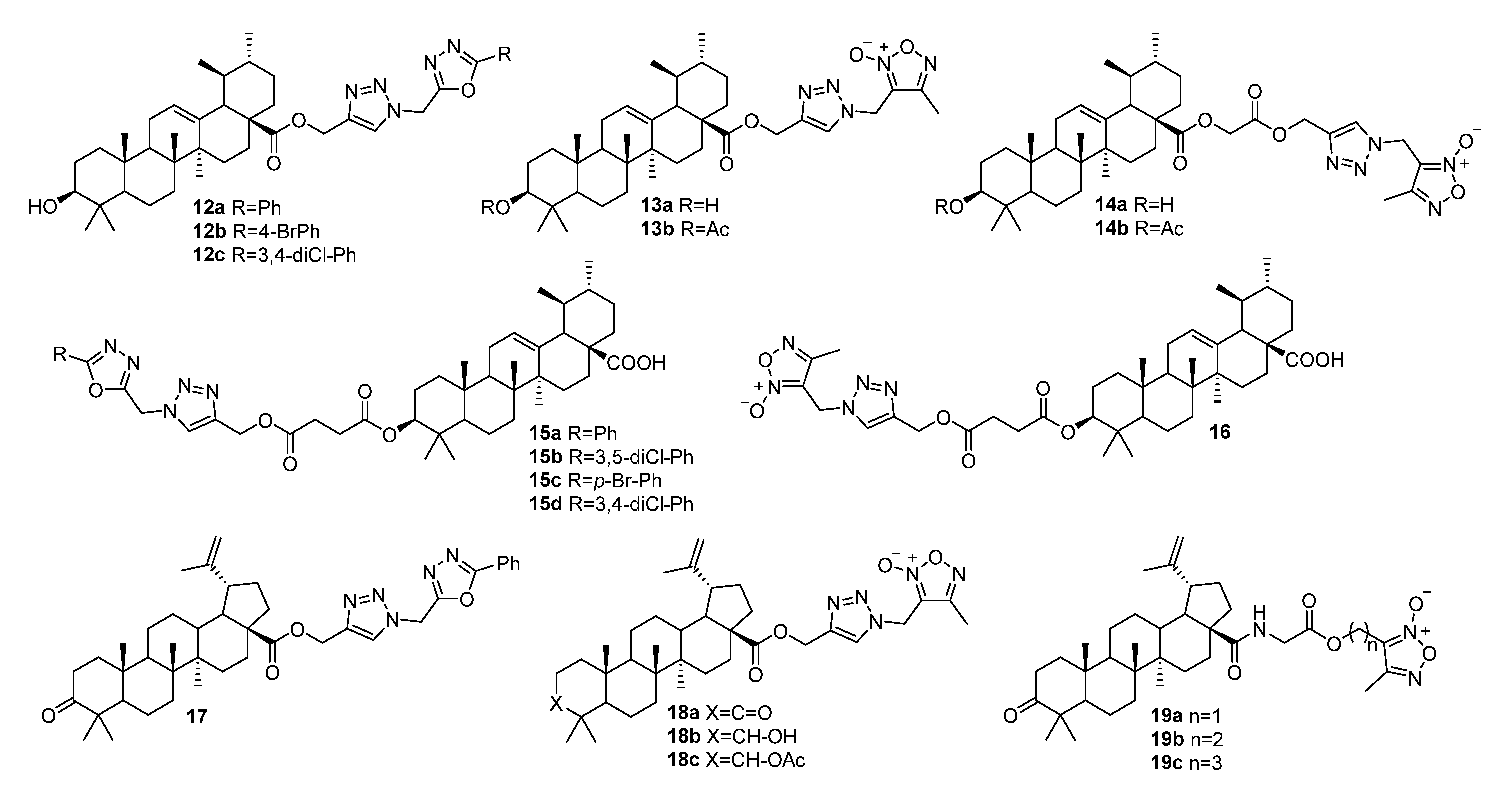
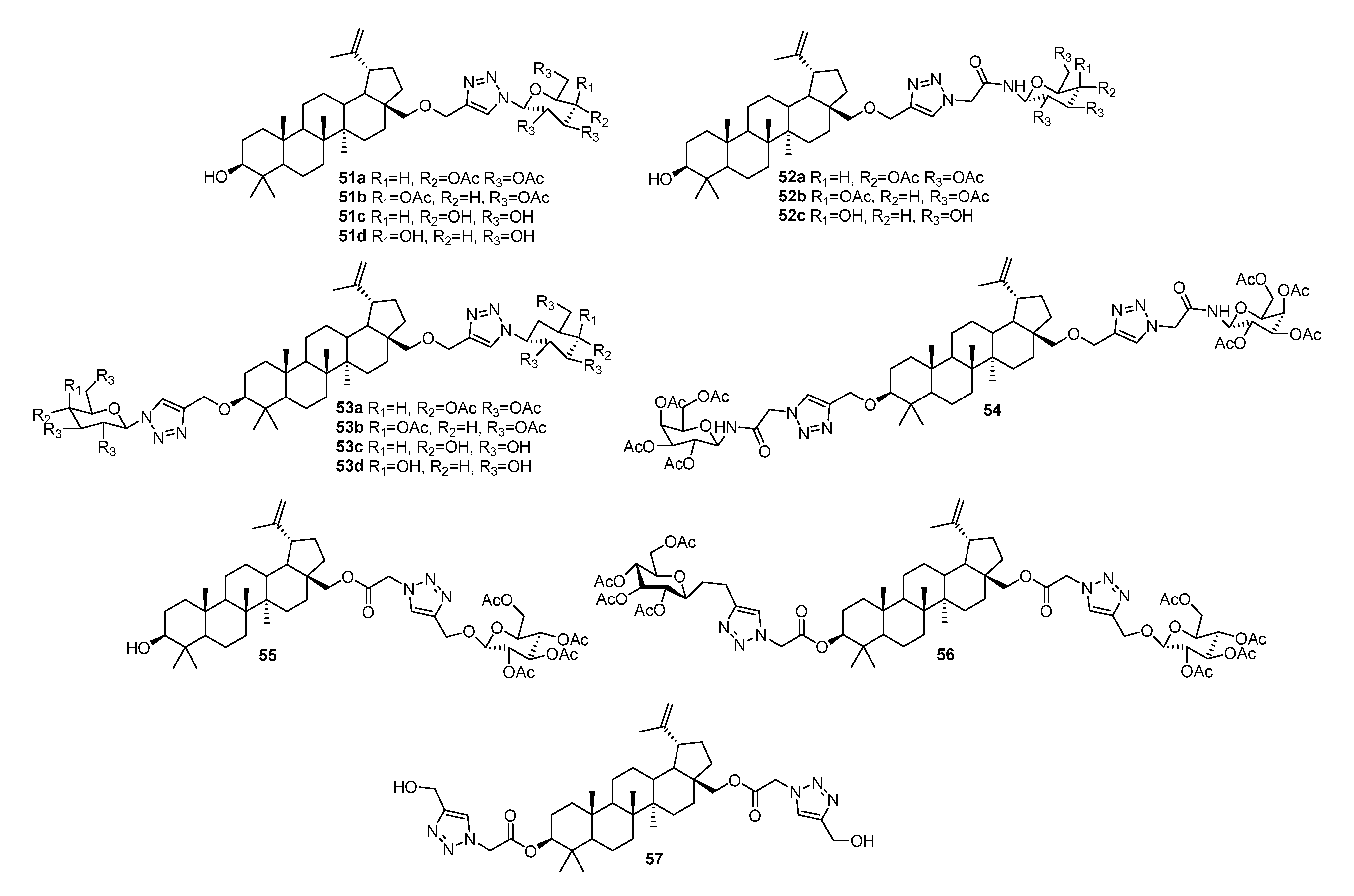
3.2. Modifications at C3 Positions
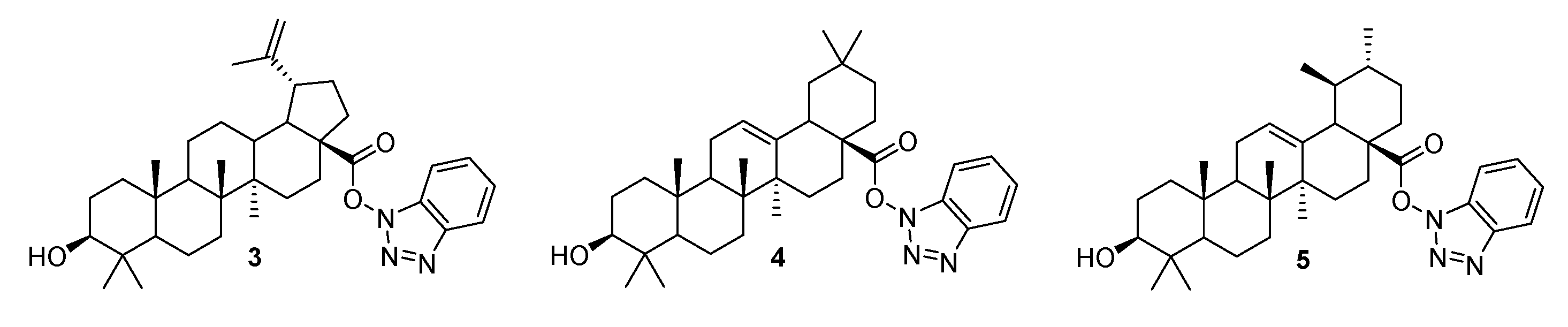
3.3. Triazoles Fused with the A-Ring
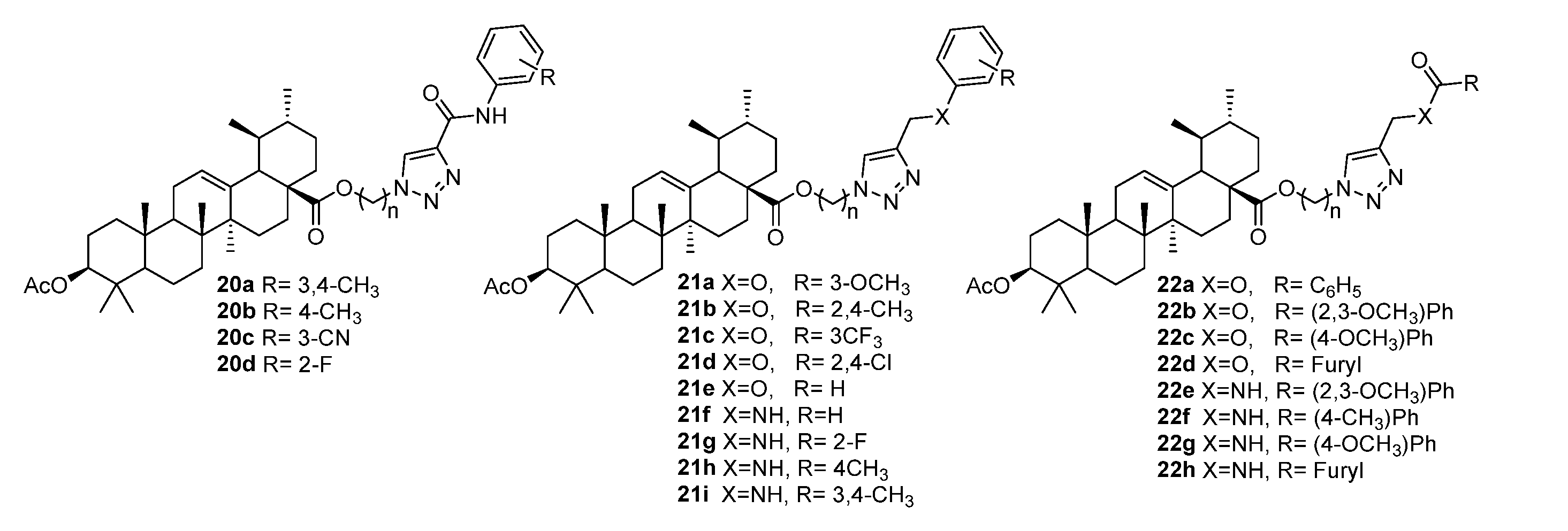
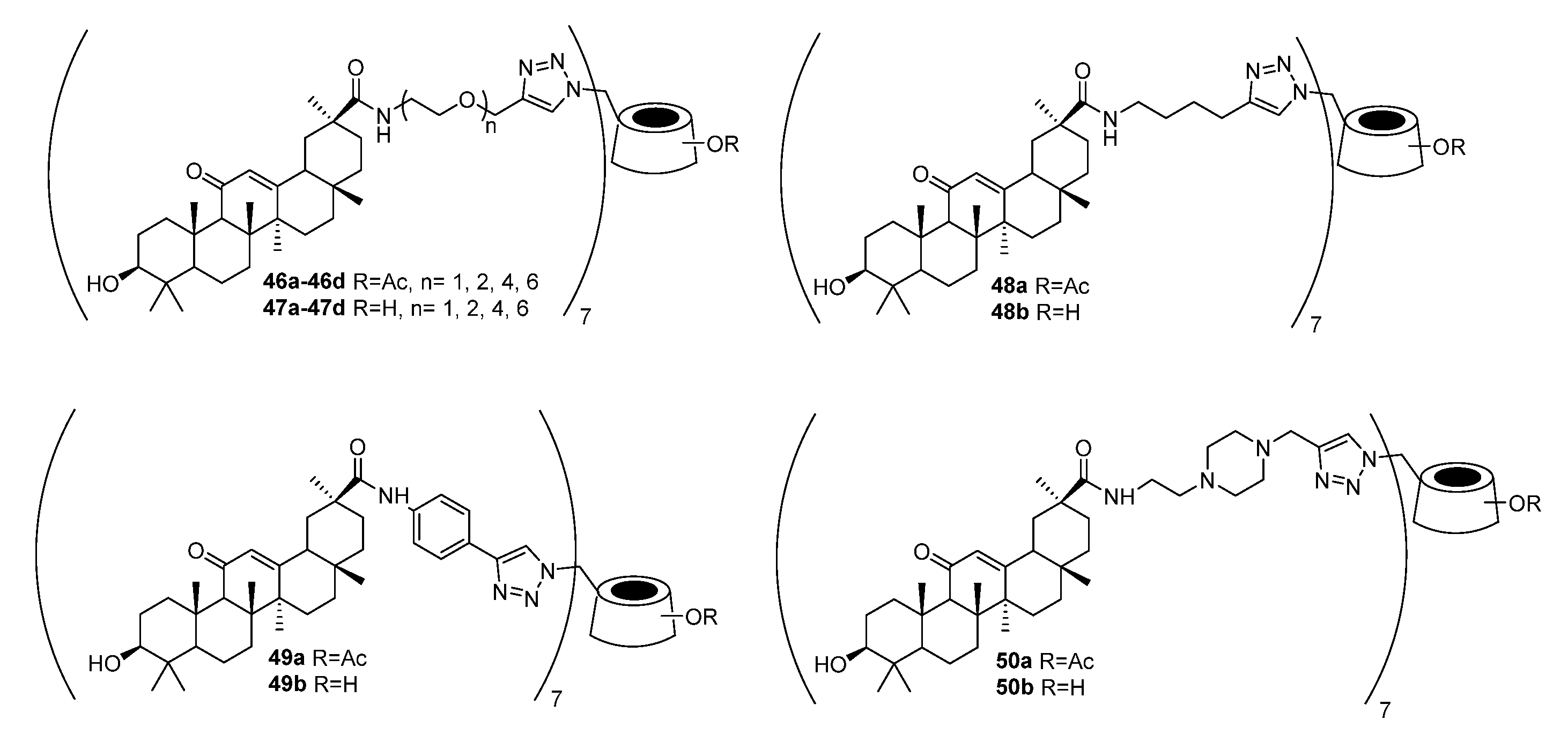
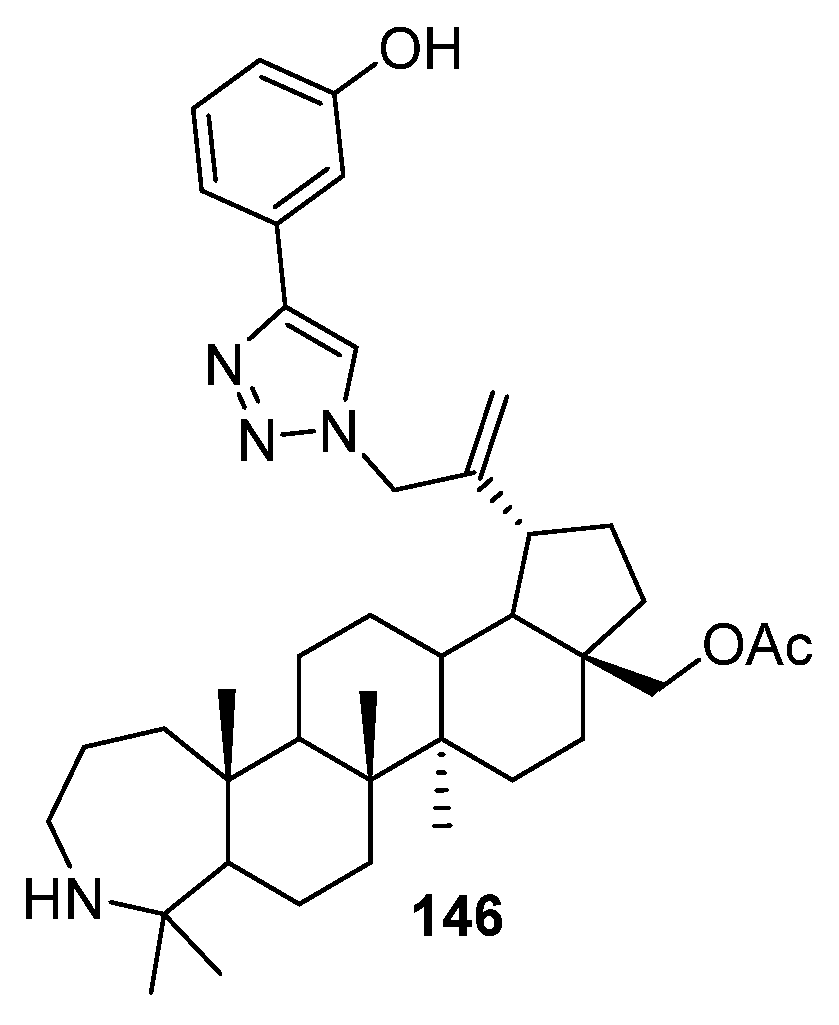
3.4. Modifications at C30, C19, and C2 Positions
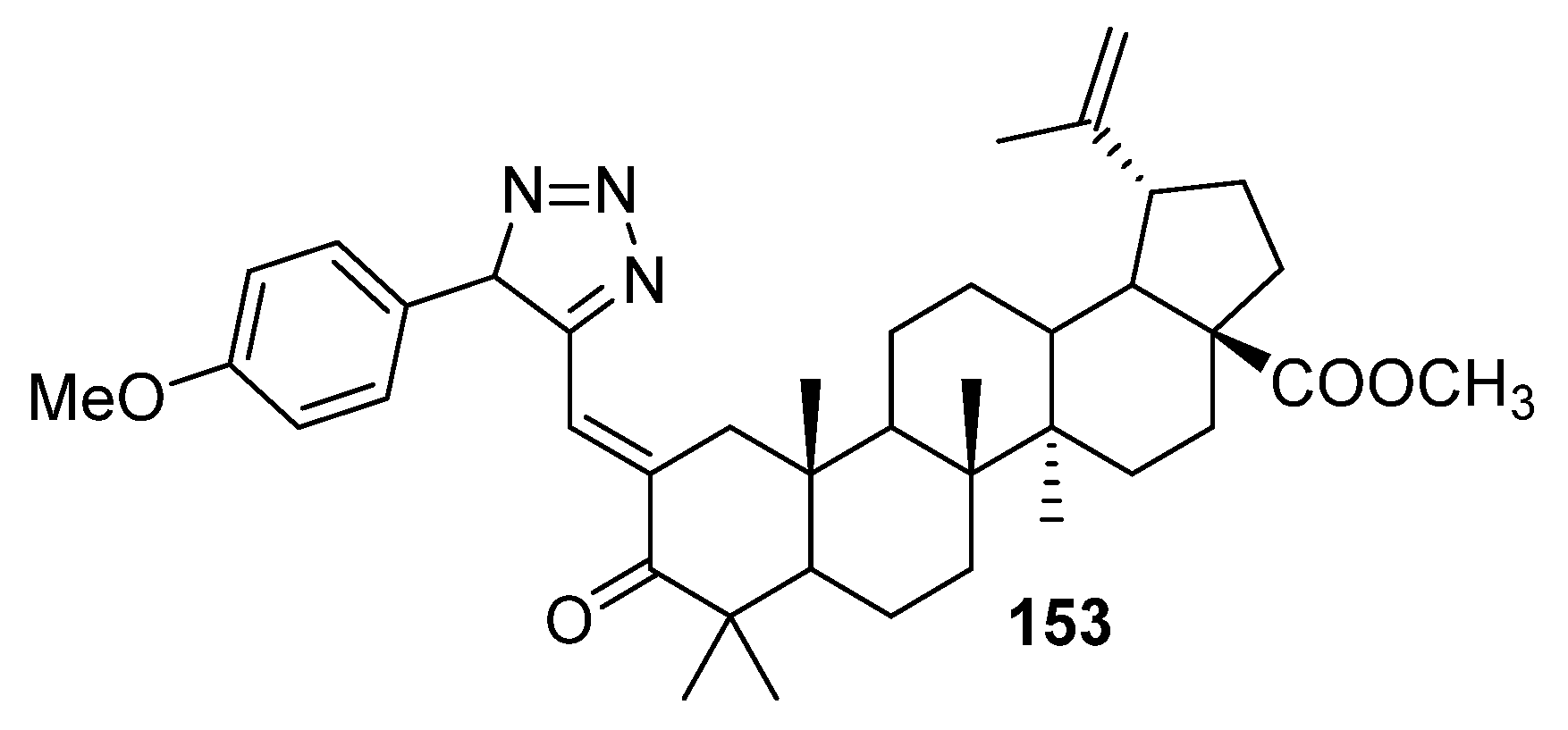
3.5. A-Seco-Triazolyl-Triterpenoids
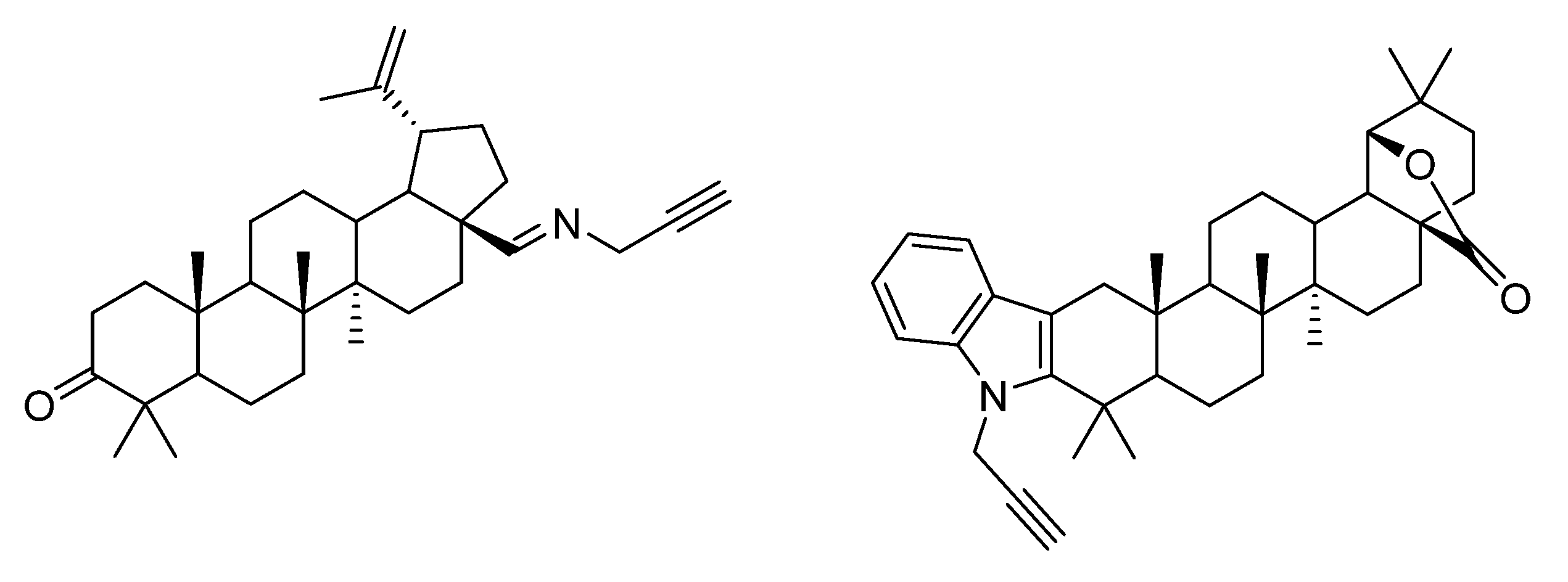
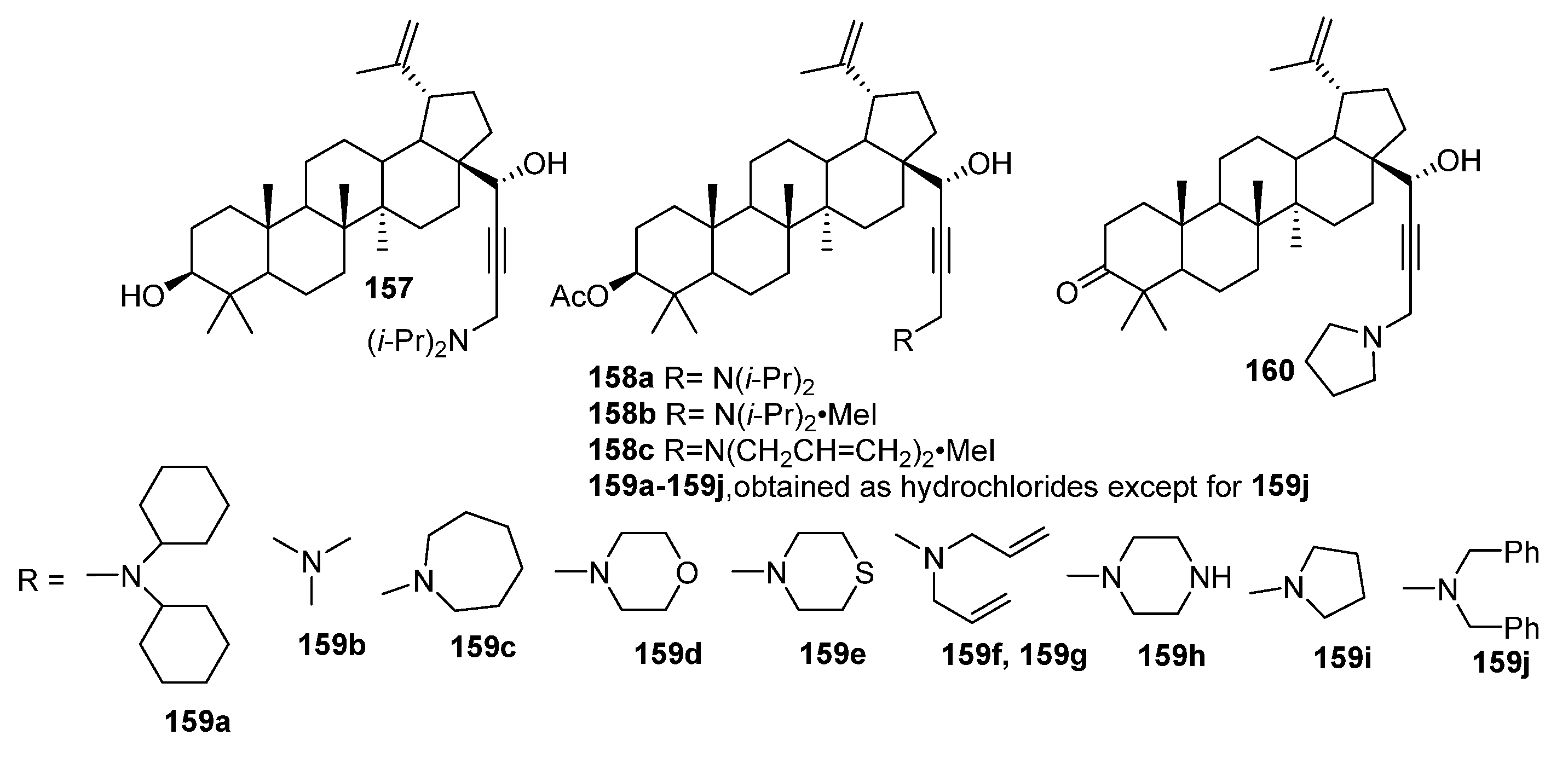
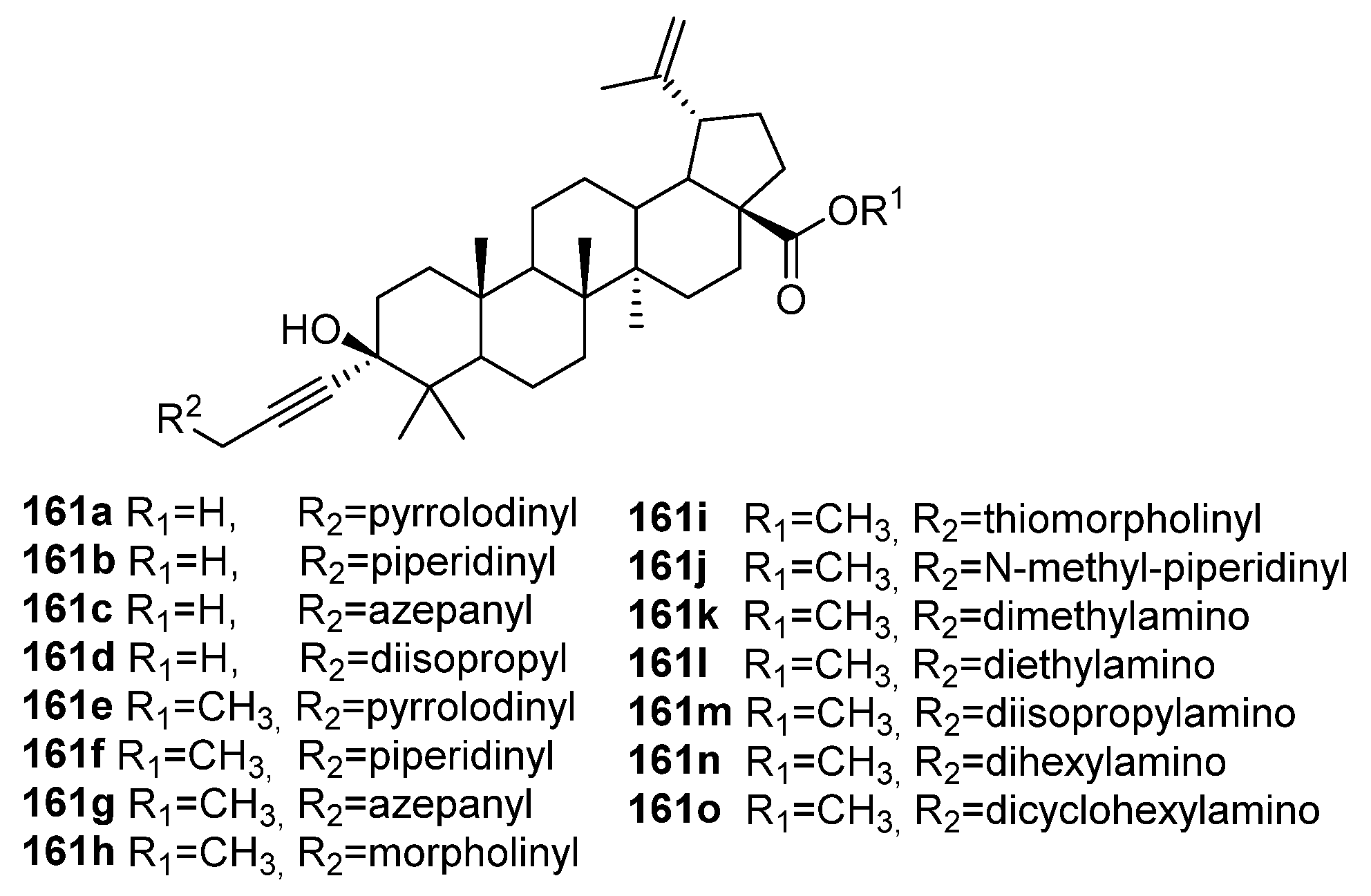
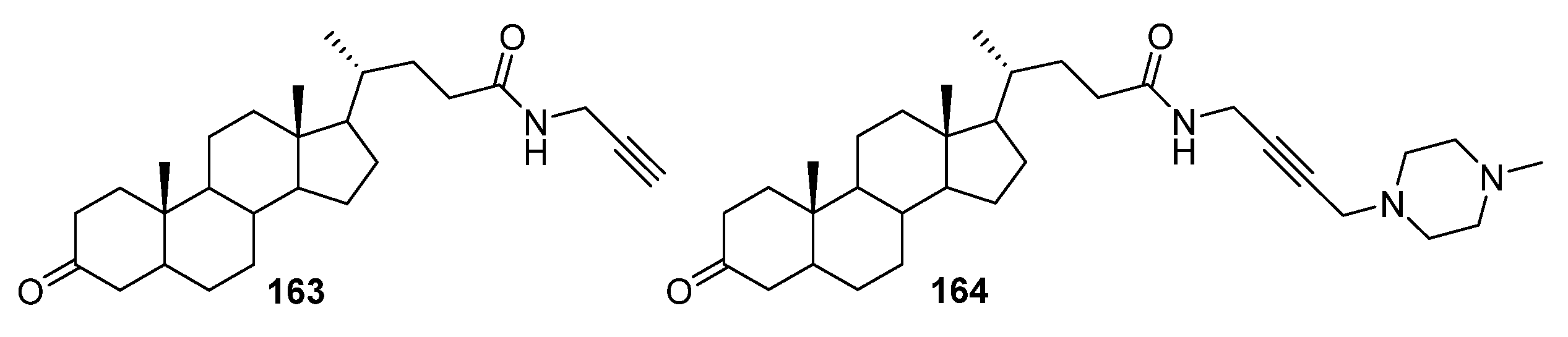
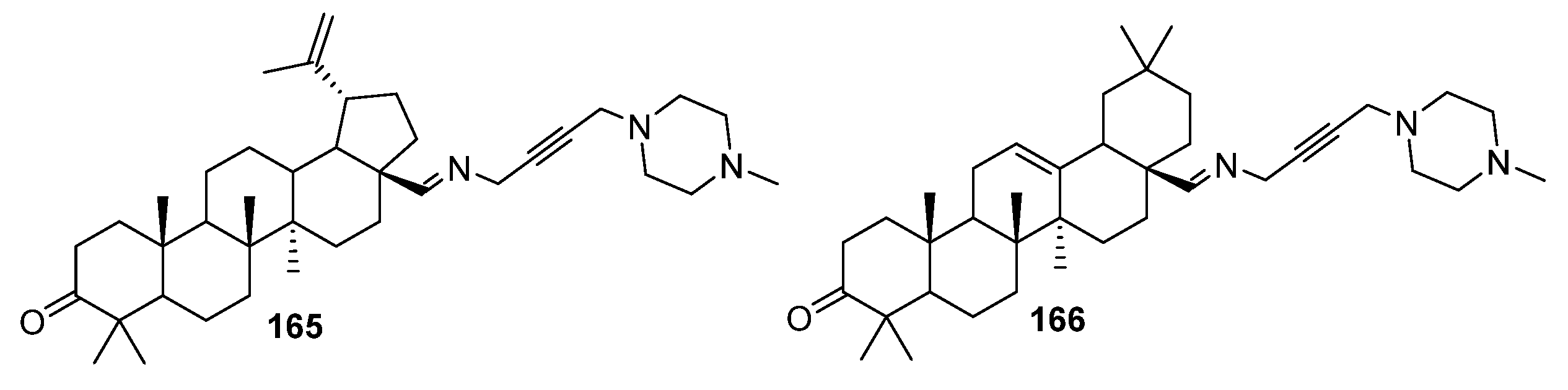
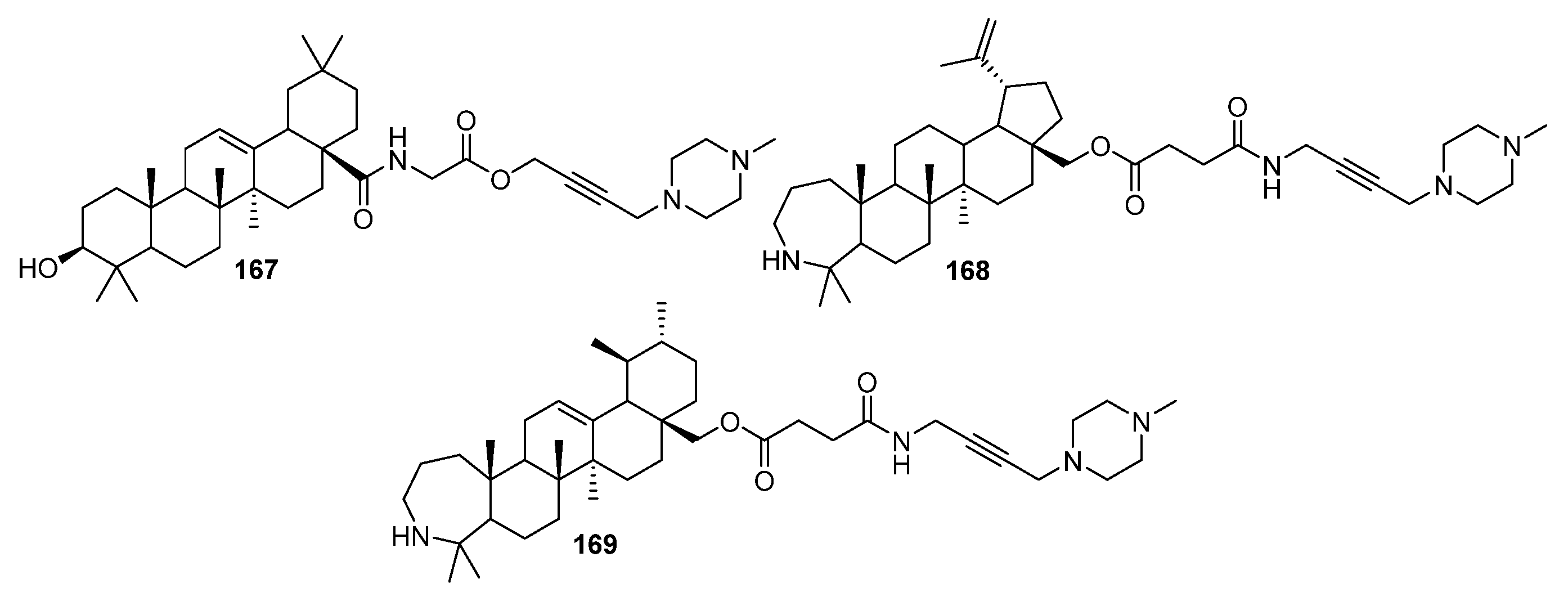
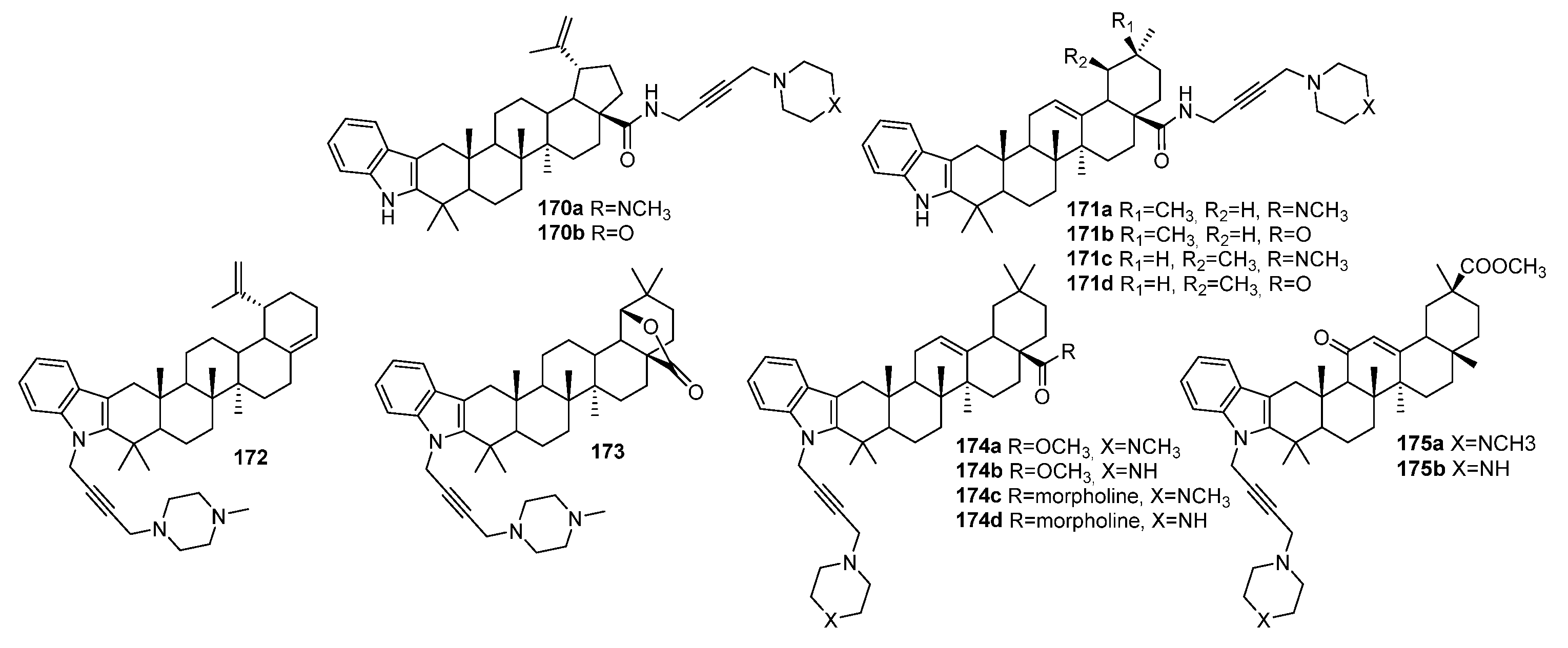
4. Modification of Triterpene Alkynes by the Mannich Reaction
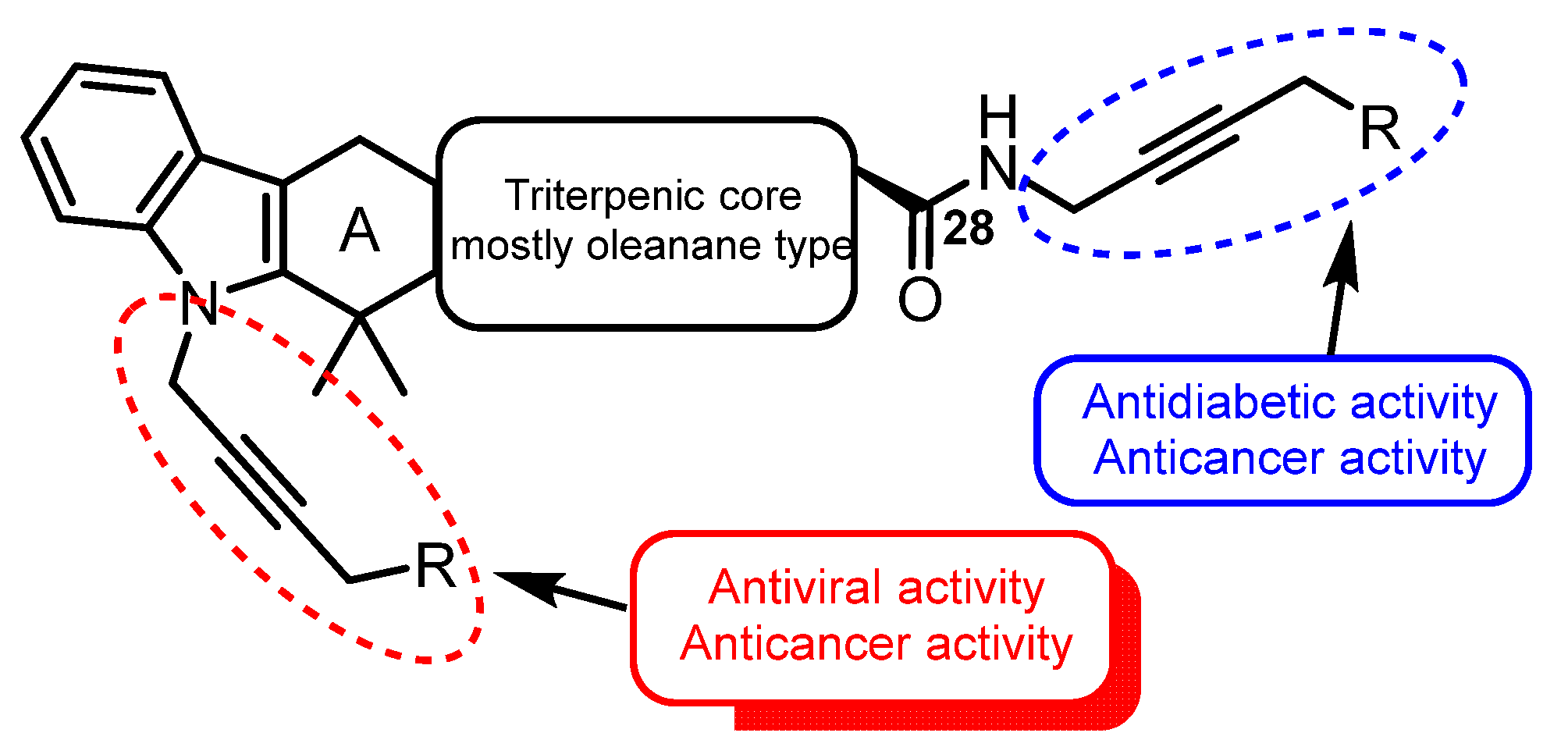
5. Pharmacological Activity
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AA | Asiatic acid |
| AZT | Azidothymidine |
| BA | Betulinic acid |
| BE | Betulin |
| CC50 | Cytotoxicity concentration |
| CuAAC | Copper-catalyzed azide-alkyne cycloaddition |
| DCC | N,N′-Dicyclohexylcarbodiimide |
| DIPEA | N,N-Diisopropylethylamine |
| DIEA | N,N-Diisopropylethylamine |
| DMAP | 4-Dimethylaminopyridine |
| DMF | Dimethylformamide |
| EDCI | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide |
| GA | Glycyrrhetinic acid |
| GTP | Guanosine triphosphate |
| HATU | Hexafluorophosphate azabenzotriazole tetramethyl uronium |
| HCMV | Human cytomegalovirus |
| HOBt | Hydroxybenzotriazole |
| HPV-11 | Human papillomavirus 11 |
| HSV-1 | Herpes simplex virus 1 |
| IC50 | Half maximal inhibitory concentration |
| MA | Maslinic acid |
| MoA | Morolic acid |
| NBS | N-Bromosuccinimide |
| OA | Oleanolic acid |
| POE | Polyoxyethylene |
| PTP1B | Protein-tyrosine phosphatase 1B |
| RSV | respiratory syncytial virus |
| rtPCR | reverse transcription polymerase chain reaction |
| TEA | Tetraethylammonium |
| UA | Ursolic acid |
| β-CD | β-Cyclodextrin |
| β-KBA | β-11-keto-boswellic acid |
References
- Jiang, X.; Hao, X.; Jing, L.; Wu, G.; Kang, D.; Liu, X.; Zhan, P. Recent applications of click chemistry in drug discovery. Expert. Opin. Drug Discov. 2019, 14, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.M. 1,2,3-Triazole hybrids as anticancer agents: A review. Arch. Pharm. 2022, 355, e2100158. [Google Scholar] [CrossRef] [PubMed]
- Roman, G. Mannich bases in medicinal chemistry and drug design. Eur. J. Med. Chem. 2015, 89, 743–816. [Google Scholar] [CrossRef]
- Biersack, B.; Ahmed, K.; Padhye, S.; Schobert, R. Recent developments concerning the application of the Mannich reaction for drug design. Expert. Opin. Drug Discov. 2018, 13, 39–49. [Google Scholar] [CrossRef]
- Bhilare, N.V.; Marulkar, V.S.; Shirote, P.J.; Dombe, S.A.; Pise, V.J.; Salve, P.L.; Biradar, S.M.; Yadav, V.D.; Jadhav, P.D.; Bodhe, A.A.; et al. Mannich Bases: Centrality in cytotoxic drug design. Med. Chem. 2022, 18, 735–756. [Google Scholar] [CrossRef]
- Hodon, J.; Borkova, L.; Pokorny, J.; Kazakova, A.; Urban, M. Design and synthesis of pentacyclic triterpene conjugates and their use in medicinal research. Eur. J. Med. Chem. 2019, 182, 111653. [Google Scholar] [CrossRef]
- Pokorny, J.; Borkova, L.; Urban, M. Click reactions in chemistry of triterpenes—Advances towards development of potential therapeutics. Curr. Med. Chem. 2018, 25, 636–658. [Google Scholar] [CrossRef]
- Csuk, R.; Deigner, H.P. The potential of click reactions for the synthesis of bioactive triterpenes. Bioorganic Med. Chem. Lett. 2019, 29, 949–958. [Google Scholar] [CrossRef]
- Kazakova, O.B.; Medvedeva, N.I.; Tolstikov, G.A.; Kukovinets, O.S.; Yamansarov, E.Y.; Spirikhin, L.V.; Gubaidullin, A.T. Synthesis of terminal acetylenes using POCl3 in pyridine as applied to natural triterpenoids. Mendeleev Commun. 2010, 20, 234–236. [Google Scholar] [CrossRef]
- Petrova, A.V.; Khusnutdinova, E.F.; Mustafin, A.G.; Kazakova, O.B. Synthesis and aminoalkylation of N-propargyl triterpene aldimines. Russ. J. Org. Chem. 2020, 56, 174–176. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Petrova, A.V.; Bashirova, G.M.; Kazakova, O.B. N-Propargylation of indolo-triterpenoids and their application in Mannich reaction. Molbank 2019, 2019, M1065. [Google Scholar] [CrossRef]
- Petrova, A.; Tretyakova, E.; Khusnutdinova, E.; Kazakova, O.; Slita, A.; Zarubaev, V.; Ma, X.; Jin, H.; Xu, H.; Xiao, S. Antiviral opportunities of Mannich bases derived from triterpenic N-propargylated indoles. Chem. Biol. Drug Des. 2024, 103, e14370. [Google Scholar] [CrossRef] [PubMed]
- da Silva, E.F.; Antunes Fernandes, K.H.; Diedrich, D.; Gotardi, J.; Freire Franco, M.S.; Tomich de Paula da Silva, C.H.; Duarte de Souza, A.P.; Baggio Gnoatto, S.C. New triazole-substituted triterpene derivatives exhibiting anti-RSV activity: Synthesis, biological evaluation, and molecular modeling. Beilstein J. Org. Chem. 2022, 18, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Mioc, M.; Mioc, A.; Prodea, A.; Milan, A.; Balan-Porcarasu, M.; Racoviceanu, R.; Ghiulai, R.; Iovanescu, G.; Macasoi, I.; Draghici, G.; et al. Novel triterpenic acid-benzotriazole esters act as pro-apoptotic antimelanoma agents. Int. J. Mol. Sci. 2022, 23, 9992. [Google Scholar] [CrossRef]
- Murlykina, M.; Pavlovska, T.; Semenenko, O.; Kolomiets, O.; Sanin, E.; Morozova, A.; Kornet, M.; Musatov, V.; Kulyk, K.; Mazepa, A.; et al. Effective three-step construction of betulonic acid hybrids with heterocycle-containing peptidomimetic fragments. Chem. Sel. 2023, 8, e202301250. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Jastrzębska, M.; Marciniec, K.; Chrobak, E.; Bębenek, E.; Latocha, M.; Kuśmierz, D.; Boryczka, S. Design, synthesis and biological activity of 1,4-quinone moiety attached to betulin derivatives as potent DT-diaphorase substrate. Bioorganic Chem. 2021, 106, 104478. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Jastrzębska, M.; Marciniec, K.; Chrobak, E.; Bębenek, E.; Boryczka, S. Lipophilicity, pharmacokinetic properties, and molecular docking study on SARS-CoV-2 target for betulin triazole derivatives with attached 1,4-quinone. Pharmaceutics 2021, 13, 781. [Google Scholar] [CrossRef]
- Popov, S.A.; Semenova, M.D.; Baev, D.S.; Frolova, T.S.; Shestopalov, M.A.; Wang, C.; Qi, Z.; Shults, E.E.; Turks, M. Synthesis and cytotoxicity of hybrids of 1,3,4- or 1,2,5-oxadiazoles tethered from ursane and lupane core with 1,2,3-triazole. Steroids 2020, 162, 108698. [Google Scholar] [CrossRef]
- Liu, Z.; Bai, X.; Sheng, L.; Sun, L.; Li, J.; Zhang, T. Ursolic acid derivatives bearing 1, 2, 3-triazole moieties as potential PTP1B Inhibitors. Chem. Nat. Compnd. 2023, 59, 508–511. [Google Scholar] [CrossRef]
- Luan, T.; Jin, C.; Jin, C.M.; Gong, G.H.; Quan, Z.S. Synthesis and biological evaluation of ursolic acid derivatives bearing triazole moieties as potential anti-Toxoplasma gondii agents. J. Enzym. Inhib. Med. Chem. 2019, 34, 761–772. [Google Scholar] [CrossRef]
- Zhang, T.Y.; Li, C.S.; Cao, L.T.; Bai, X.Q.; Zhao, D.H.; Sun, S.M. New ursolic acid derivatives bearing 1,2,3-triazole moieties: Design, synthesis and anti-inflammatory activity in vitro and in vivo. Mol. Divers. 2022, 26, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Yang, G.; Deng, T.; Wang, J.; Zhou, H.; Popov, S.A.; Shults, E.E.; Wang, C. Design and linkage optimization of ursane-thalidomide-based PROTACs and identification of their targeted-degradation properties to MDM2 protein. Bioorganic Chem. 2021, 111, 104901. [Google Scholar] [CrossRef] [PubMed]
- Kapkoti, D.S.; Kumar, S.; Kumar, A.; Darokar, M.P.; Pal, A.; Bhakuni, R.S. Design and synthesis of novel glycyrrhetinic acid-triazole derivatives that exert anti-plasmodial activity inducing mitochondrial-dependent apoptosis in Plasmodium falciparum. New J. Chem. 2023, 47, 6967–6982. [Google Scholar] [CrossRef]
- Bian, M.; Zhen, D.; Shen, Q.K.; Du, H.H.; Ma, Q.Q.; Quan, Z.S. Structurally modified glycyrrhetinic acid derivatives as anti-inflammatory agents. Bioorganic Chem. 2021, 107, 104598. [Google Scholar] [CrossRef]
- Rehman, N.U.; Ullah, S.; Alam, T.; Halim, S.A.; Mohanta, T.K.; Khan, A.; Anwar, M.U.; Csuk, R.; Avula, S.K.; Al-Harrasi, A. Discovery of new boswellic acid hybrid 1H-1,2,3-triazoles for diabetic management: In vitro and in silico studies. Pharmaceuticals 2023, 16, 229. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Ma, X.; Liang, S.; Gao, Q.; Tretyakova, E.V.; Zhang, Y.; Zhou, D.; Xiao, S. Facial Synthesis and bioevaluation of well-defined OEGylated betulinic acid-cyclodextrin conjugates for inhibition of influenza infection. Molecules 2022, 27, 1163. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, X.; Zhu, Y.; Si, L.; Zhang, B.; Zhang, Y.; Zhang, L.; Zhou, D.; Xiao, S. Synthesis of a hexavalent betulinic acid derivative as a hemagglutinin-targeted influenza virus entry inhibitor. Mol. Pharm. 2020, 17, 2546–2554. [Google Scholar] [CrossRef]
- Liang, S.; Li, M.; Yu, X.; Jin, H.; Zhang, Y.; Zhang, L.; Zhou, D.; Xiao, S. Synthesis and structure-activity relationship studies of water-soluble β-cyclodextrin-glycyrrhetinic acid conjugates as potential anti-influenza virus agents. Eur. J. Med. Chem. 2019, 166, 328–338. [Google Scholar] [CrossRef]
- Liang, S.; Ma, X.; Li, M.; Yi, Y.; Gao, Q.; Zhang, Y.; Zhang, L.; Zhou, D.; Xiao, S. Novel β-cyclodextrin-based heptavalent glycyrrhetinic acid conjugates: Synthesis, characterization, and anti-influenza activity. Front. Chem. 2022, 10, 836955. [Google Scholar] [CrossRef]
- Grymel, M.; Pastuch-Gawołek, G.; Lalik, A.; Zawojak, M.; Boczek, S.; Krawczyk, M.; Erfurt, K. Glycoconjugation of betulin derivatives using copper-catalyzed 1,3-dipolar azido-alkyne cycloaddition reaction and a preliminary assay of cytotoxicity of the obtained compounds. Molecules 2020, 25, 6019. [Google Scholar] [CrossRef]
- Hodon, J.; Frydrych, I.; Trhlikova, Z.; Pokorny, J.; Borkova, L.; Benicka, S.; Vlk, M.; Liskova, B.; Kubickova, A.; Medvedikova, M.; et al. Triterpenoid pyrazines and pyridines—Synthesis, cytotoxicity, mechanism of action, preparation of prodrugs. Eur. J. Med. Chem. 2022, 243, 114777. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, E.F.; Petrova, A.V.; Faskhutdinova, L.N.; Kukovinets, O.S. 1,2,3-Triazole derivatives based on glycine and phenylalanine amides and triterpene acids. Russ. J. Org. Chem. 2018, 54, 639–643. [Google Scholar] [CrossRef]
- Su, Y.; Meng, L.; Sun, J.; Li, W.; Shao, L.; Chen, K.; Zhou, D.; Yang, F.; Yu, F. Design, synthesis of oleanolic acid-saccharide conjugates using click chemistry methodology and study of their anti-influenza activity. Eur. J. Med. Chem. 2019, 182, 111622. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Su, Y.; Zhang, Y.; Yang, F.; Zhang, J.; Tang, T.; Yu, F. Nine-valent oleanolic acid conjugates as potent inhibitors blocking the entry of influenza A virus. Eur. J. Med. Chem. 2023, 258, 115562. [Google Scholar] [CrossRef]
- Lv, M.; Qian, X.; Li, S.; Gong, J.; Wang, Q.; Qian, Y.; Su, Z.; Xue, X.; Liu, H.K. Unlocking the potential of iridium and ruthenium arene complexes as anti-tumor and anti-metastasis chemotherapeutic agents. J. Inorg. Biochem. 2023, 238, 112057. [Google Scholar] [CrossRef]
- Huang, R.Z.; Liang, G.B.; Li, M.S.; Fang, Y.L.; Zhao, S.F.; Zhou, M.M.; Liao, Z.X.; Sun, J.; Wang, H.S. Synthesis and discovery of asiatic acid based 1,2,3-triazole derivatives as antitumor agents blocking NF-κB activation and cell migration. MedChemComm 2019, 10, 584–597. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, W.; Zhang, Y.; Fu, X.; Ping, K.; Zhao, J.; Lei, Y.; Mou, Y.; Wang, S. Synthesis and biological evaluation of celastrol derivatives as potential anti-glioma agents by activating RIP1/RIP3/MLKL pathway to induce necroptosis. Eur. J. Med. Chem. 2022, 229, 114070. [Google Scholar] [CrossRef]
- Semenova, M.D.; Popov, S.A.; Sorokina, I.V.; Meshkova, Y.V.; Baev, D.S.; Tolstikova, T.G.; Shults, E.E. Conjugates of lupane triterpenoids with arylpyrimidines: Synthesis and anti-inflammatory activity. Steroids 2022, 184, 109042. [Google Scholar] [CrossRef]
- Chen, Z.; Jiang, Y.; Xu, C.; Sun, X.; Ma, C.; Xia, Z.; Zhao, H. Oleanane-type triterpene conjugates with 1H-1,2,3-triazole possessing of fungicidal activity. Molecules 2022, 27, 4928. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, J.; Min, X.; Liang, B.; Sun, J.; Lin, K.; Xiong, Z.; Xu, X.; Chen, W.-H. 1, 2, 3-Triazole-based betulinic acid derivatives as α-glucosidase inhibitors: Synthesis and in vitro and in vivo biological evaluation. J. Mol. Struct. 2024, 1310, 138294. [Google Scholar] [CrossRef]
- Lahmadi, G.; Horchani, M.; Dbeibia, A.; Mahdhi, A.; Romdhane, A.; Lawson, A.M.; Daich, A.; Harrath, A.H.; Ben Jannet, H.; Othman, M. Novel oleanolic acid-phtalimidines tethered 1,2,3 triazole hybrids as promising antibacterial agents: Design, synthesis, in vitro experiments and in silico docking studies. Molecules 2023, 28, 4655. [Google Scholar] [CrossRef] [PubMed]
- Avula, S.K.; Rehman, N.U.; Khan, F.; Ullah, O.; Halim, S.A.; Khan, A.; Al-Harrasi, A. Triazole-tethered boswellic acid derivatives against breast cancer: Synthesis, in vitro, and in-silico studies. J. Mol. Struct. 2023, 1282, 135181. [Google Scholar] [CrossRef]
- Li, N.; Song, J.; Li, D. Synthesis and Antiproliferative Activity of Ester Derivatives of Mogrol through JAK2/STAT3 Pathway. Chem. Biodivers 2022, 19, e202100742. [Google Scholar] [CrossRef]
- Pavley, Y.R.; Yamansarov, E.Y.; Evteev, S.A.; Lopatukhina, E.V.; Zyk, N.V.; Erofeev, A.S.; Beloglazkina, E.K. New acetylenic derivatives of bile acids as versatile precursors for the preparation of prodrugs. Synthesis and cytotoxicity study. Russ. Chem. Bull. 2023, 72, 724–739. [Google Scholar] [CrossRef]
- Boulila, B.; Horchani, M.; Duval, R.; Othman, M.; Daich, A.; Jannet, H.B.; Lawson, A.M. Design, semi-synthesis and molecular docking of new antibacterial and antibiofilm triazole conjugates from hydroxy-triterpene acids and fluoroquinolones. New J. Chem. 2023, 47, 15973–15986. [Google Scholar] [CrossRef]
- Ozdemir, Z.; Rybkova, M.; Vlk, M.; Saman, D.; Rarova, L.; Wimmer, Z. Synthesis and pharmacological effects of diosgenin-betulinic acid conjugates. Molecules 2020, 25, 3546. [Google Scholar] [CrossRef]
- Ozdemir, Z.; Saman, D.; Bednarova, L.; Pazderkova, M.; Janovska, L.; Nonappa; Wimmer, Z. Aging-induced structural transition of nanoscale oleanolic acid amphiphiles and selectivity against Gram-positive bacteria. ACS Appl. Nano Mater. 2022, 5, 3799–3810. [Google Scholar] [CrossRef]
- Ozdemir, Z.; Saman, D.; Bertula, K.; Lahtinen, M.; Bednárová, L.; Pazderková, M.; Rárová, L.; Nonappa; Wimmer, Z. Rapid self-healing and thixotropic organogelation of amphiphilic oleanolic acid-spermine conjugates. Langmuir 2021, 37, 2693–2706. [Google Scholar] [CrossRef]
- Bildziukevich, U.; Ozdemir, Z.; Saman, D.; Vlk, M.; Slouf, M.; Rarova, L.; Wimmer, Z. Novel cytotoxic 1,10-phenanthroline-triterpenoid amphiphiles with supramolecular characteristics capable of coordinating 64Cu(II) labels. Org. Biomol. Chem. 2022, 20, 8157–8163. [Google Scholar] [CrossRef]
- Ozdemir, Z.; Bildziukevich, U.; Capkova, M.; Lovecka, P.; Rarova, L.; Saman, D.; Zgarbova, M.; Lapunikova, B.; Weber, J.; Kazakova, O.; et al. Triterpenoid-PEG ribbons targeting selectivity in pharmacological effects. Biomedicines 2021, 9, 951. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Hu, H.; Persoons, L.; Daelemans, D.; De Jonghe, S.; Luyten, W.; Krasniqi, B.; Dehaen, W. Antibacterial and antitumoral properties of 1,2,3-triazolo fused triterpenes and their mechanism of inhibiting the proliferation of HL-60 cells. Eur. J. Med. Chem. 2021, 224, 113727. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Krasniqi, B.; Li, Y.; Dehaen, W. Triphenylphosphonium-linked derivative of allobetulin: Preparation, anticancer properties and their mechanism of inhibiting SGC-7901 cells proliferation. Bioorganic Chem. 2022, 126, 105853. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, G.; Hodon, J.; Kazakova, A.; D’Acunto, C.W.; Kanovsky, P.; Urban, M.; Strnad, M. Novel pentacyclic triterpenes exhibiting strong neuroprotective activity in SH-SY5Y cells in salsolinol- and glutamate-induced neurodegeneration models. Eur. J. Med. Chem. 2021, 213, 113168. [Google Scholar] [CrossRef] [PubMed]
- Nistor, G.; Mioc, M.; Mioc, A.; Balan-Porcarasu, M.; Racoviceanu, R.; Prodea, A.; Milan, A.; Ghiulai, R.; Semenescu, A.; Dehelean, C.; et al. The C30-modulation of betulinic acid using 1,2,4-triazole: A promising strategy for increasing its antimelanoma cytotoxic potential. Molecules 2022, 27, 7807. [Google Scholar] [CrossRef]
- Nistor, G.; Mioc, A.; Mioc, M.; Balan-Porcarasu, M.; Ghiulai, R.; Racoviceanu, R.; Avram, S.; Prodea, A.; Semenescu, A.; Milan, A.; et al. Novel Semisynthetic betulinic acid−triazole hybrids with in vitro antiproliferative potential. Processes 2023, 11, 101. [Google Scholar] [CrossRef]
- Bebenek, E.; Kadela-Tomanek, M.; Chrobak, E.; Latocha, M. 3′-[4-({[3β, 28-Bis(acetyloxy)lup-20 (29)-en-30-yl]oxy}carbonyl)-1H-1,2,3-triazol-1-yl]-3′-deoxythymidine. Molbank 2022, 2022, M1370. [Google Scholar] [CrossRef]
- Bebenek, E.; Kadela-Tomanek, M.; Chrobak, E.; Jastrzebska, M.; Ksiazek, M. Synthesis and structural characterization of a new 1,2,3-triazole derivative of pentacyclic triterpene. Crystals 2022, 12, 422. [Google Scholar] [CrossRef]
- Petrova, A.V.; Lopatina, T.V.; Mustafin, A.G.; Kazakova, O.B. Modification of azepanobetulin at the isopropenyl group. Russ. J. Org. Chem. 2020, 56, 1582–1587. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Bremond, P.; Petrova, A.V.; Kukovinets, O.S.; Kazakova, O.B. Synthesis of lupane mono-and bis-C19-(1, 2, 3-triazolyl)-triterpenoids by “Click” reaction. Lett. Org. Chem. 2017, 14, 743–747. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Petrova, A.V.; Kazakova, O.B. Antiviral potency of lupane and oleanane alkynyl-derivatives against human cytomegalovirus and papillomavirus. J. Antibiot. 2024, 77, 50–56. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, X.; Zhang, X.; Wang, J.; Li, K.; Liu, G.; Lu, K.; Zhang, X.; Xie, C.; Zheng, T.; et al. Synthesis and biological evaluation of novel allobetulon/allobetulin-nucleoside conjugates as antitumoragents. Molecules 2022, 27, 4738. [Google Scholar] [CrossRef] [PubMed]
- Sousa, J.L.C.; Albuquerque, H.M.T.; Silvestre, A.J.D.; Silva, A.M.S. Decoration of A-Ring of a lupane-type triterpenoid with different oxygen and nitrogen heterocycles. Molecules 2022, 27, 4904. [Google Scholar] [CrossRef] [PubMed]
- Kuczynska, K.; Bonczak, B.; Rarova, L.; Kvasnicova, M.; Strnad, M.; Pakulski, Z.; Cmoch, P.; Fiałkowski, M. Synthesis and cytotoxic activity of 1,2,3-triazoles derived from 2,3-seco-dihydrobetulin via a click chemistry approach. J. Mol. Struct. 2021, 1250, 131751. [Google Scholar] [CrossRef]
- Petrova, A.V. Synthesis and modification of new olenane type 2-cyano-3, 4-seco-5-alkynylderivative. AIP Conf. Proc. 2022, 2390, 020058. [Google Scholar] [CrossRef]
- Kim, J.Y.; Koo, H.M.; Kim, D.S. Development of C-20 modified betulinic acid derivatives as antitumor agents. Bioorganic Med. Chem. Lett. 2001, 11, 2405–2408. [Google Scholar] [CrossRef]
- Bieber, L.W.; da Silva, M.F. Mild and efficient synthesis of propargylamines by copper-catalyzed Mannich reaction. Tetrahedron Lett. 2004, 45, 8281–8283. [Google Scholar] [CrossRef]
- Csuk, R.; Nitsche, C.; Sczepek, R.; Schwarz, S.; Siewert, B. Synthesis of antitumor-active betulinic acid-derived hydroxypropargylamines by copper-catalyzend mannich reactions. Arch. Pharm. 2013, 346, 232–246. [Google Scholar] [CrossRef]
- Csuk, R.; Sczepek, R.; Siewert, B.; Nitsche, C. Cytotoxic betulin-derived hydroxypropargylamines trigger apoptosis. Bioorganic Med. Chem. 2013, 21, 425–435. [Google Scholar] [CrossRef]
- Govdi, A.I.; Sorokina, I.V.; Baev, D.S.; Bryzgalov, A.O.; Tolstikova, T.G.; Tolstikov, G.A.; Vasilevsky, S.F. Acetylenic derivatives of betulonic acid amide as a new type of compounds possessing spasmolytic activity. Russ. Chem. Bull. 2015, 64, 1327–1334. [Google Scholar] [CrossRef]
- Petrova, A.V.; Smirnova, I.E.; Fedij, S.V.; Pavlyukova, Y.N.; Zarubaev, V.V.; Tran Thi Phuong, T.; Myint Myint, K.; Kazakova, O.B. Synthesis and inhibition of influenza h1n1 virus by propargylaminoalkyl derivative of lithocholic acid. Molbank 2023, 2023, M1626. [Google Scholar] [CrossRef]
- Khusnutdinova, E.F.; Apryshko, G.N.; Petrova, A.V.; Kukovinets, O.S.; Kazakova, O.B. The synthesis and selective cytotoxicity of new Mannich bases, derivatives of 19- and 28-alkynyltriterpenoids. Russ. J. Bioorganic Chem. 2018, 44, 123–127. [Google Scholar] [CrossRef]
- Petrova, A.V. Synthesis and aminomethylation of A-azepane-fused uvaol and betulin derivatives. Russ. J. Org. Chem. 2023, 59, 202–206. [Google Scholar] [CrossRef]
- Luo, M.L.; Zhao, Q.; He, X.H.; Xie, X.; Zhu, H.P.; You, F.M.; Peng, C.; Zhan, G.; Huang, W. Research progress of indole-fused derivatives as allosteric modulators: Opportunities for drug development. Biomed. Pharmacother. 2023, 162, 114574. [Google Scholar] [CrossRef] [PubMed]
- Khusnutdinova, E.F.; Petrova, A.V.; Kukovinets, O.S.; Kazakova, O.B. Synthesis and cytotoxicity of 28-N-propargylaminoalkylated 2,3-indolotriterpenic acids. Nat. Prod. Commun. 2018, 13, 665–668. [Google Scholar] [CrossRef]
- Petrova, A.V.; Babkov, D.A.; Khusnutdinova, E.F.; Baikova, I.P.; Kazakova, O.B.; Sokolova, E.V.; Spasov, A.A. α-Glucosidase Inhibitors Based on Oleanolic Acid for the Treatment of Immunometabolic Disorders. Appl. Sci. 2023, 13, 9269. [Google Scholar] [CrossRef]



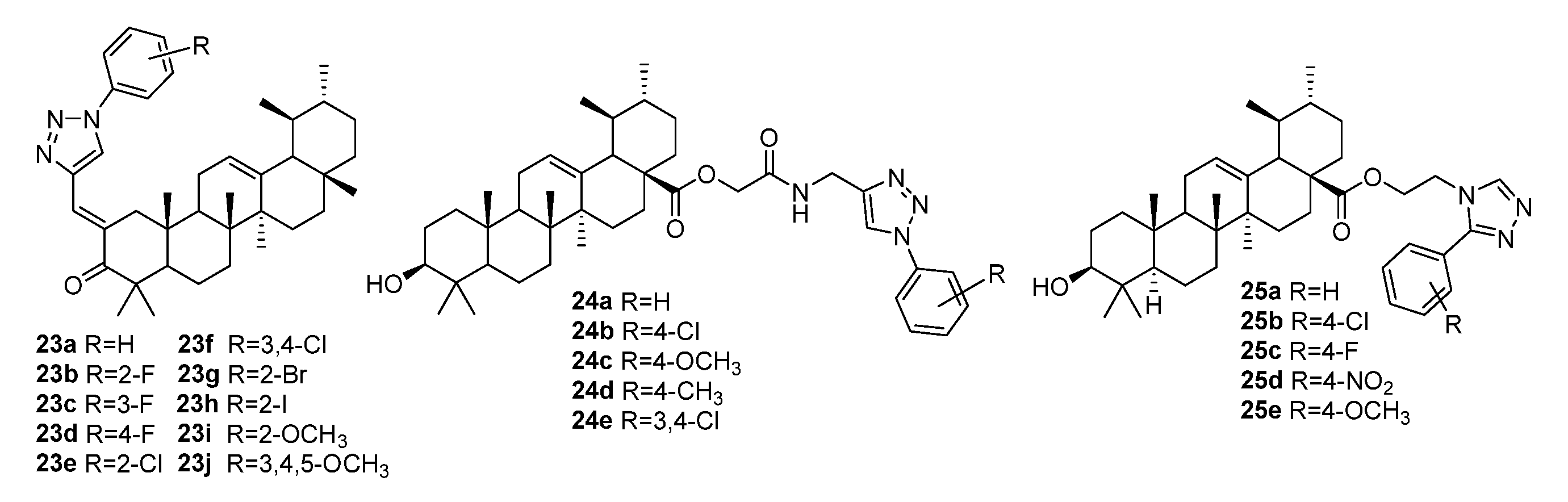
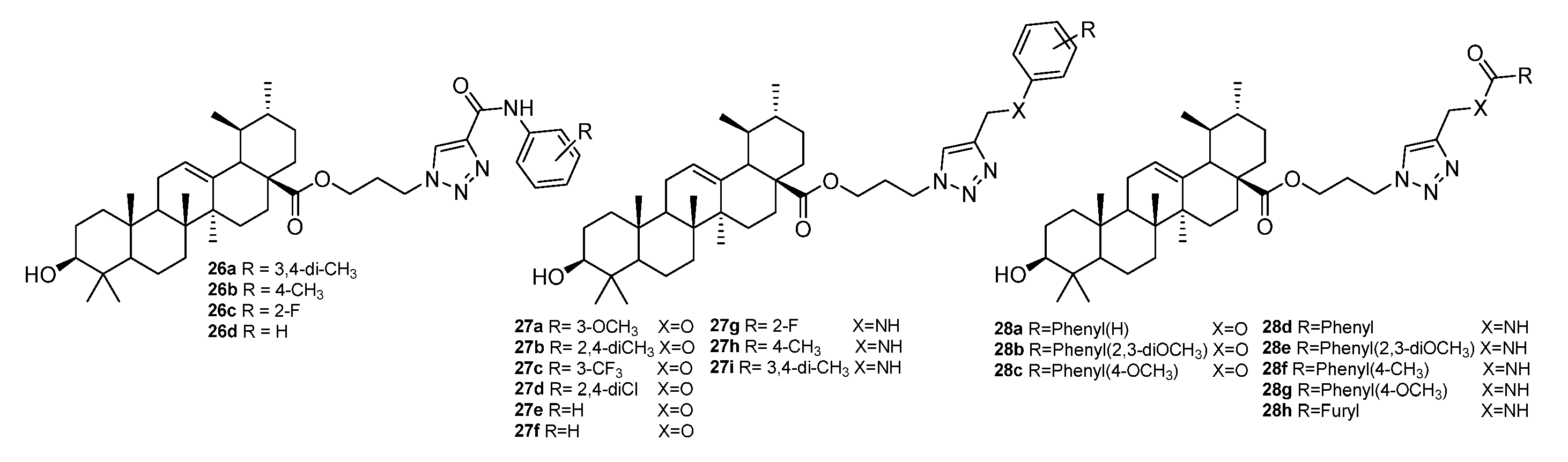
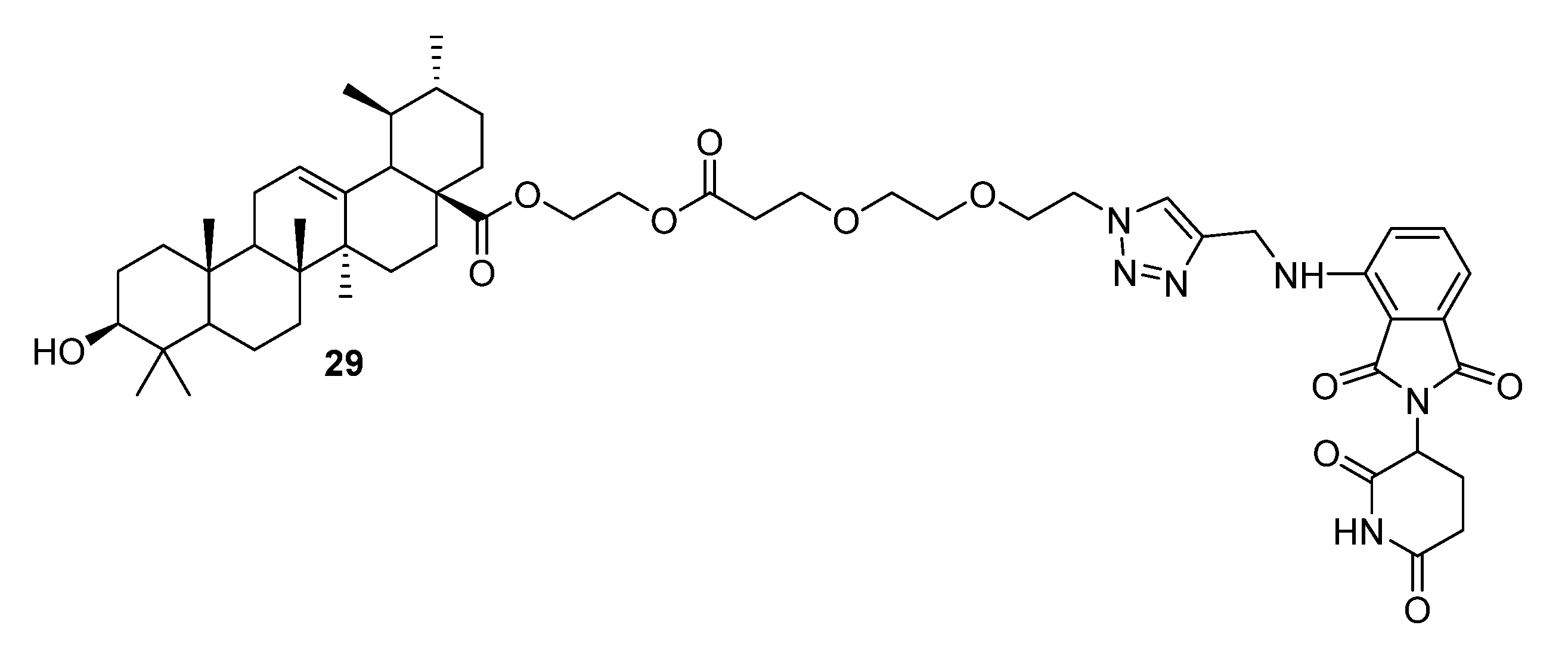
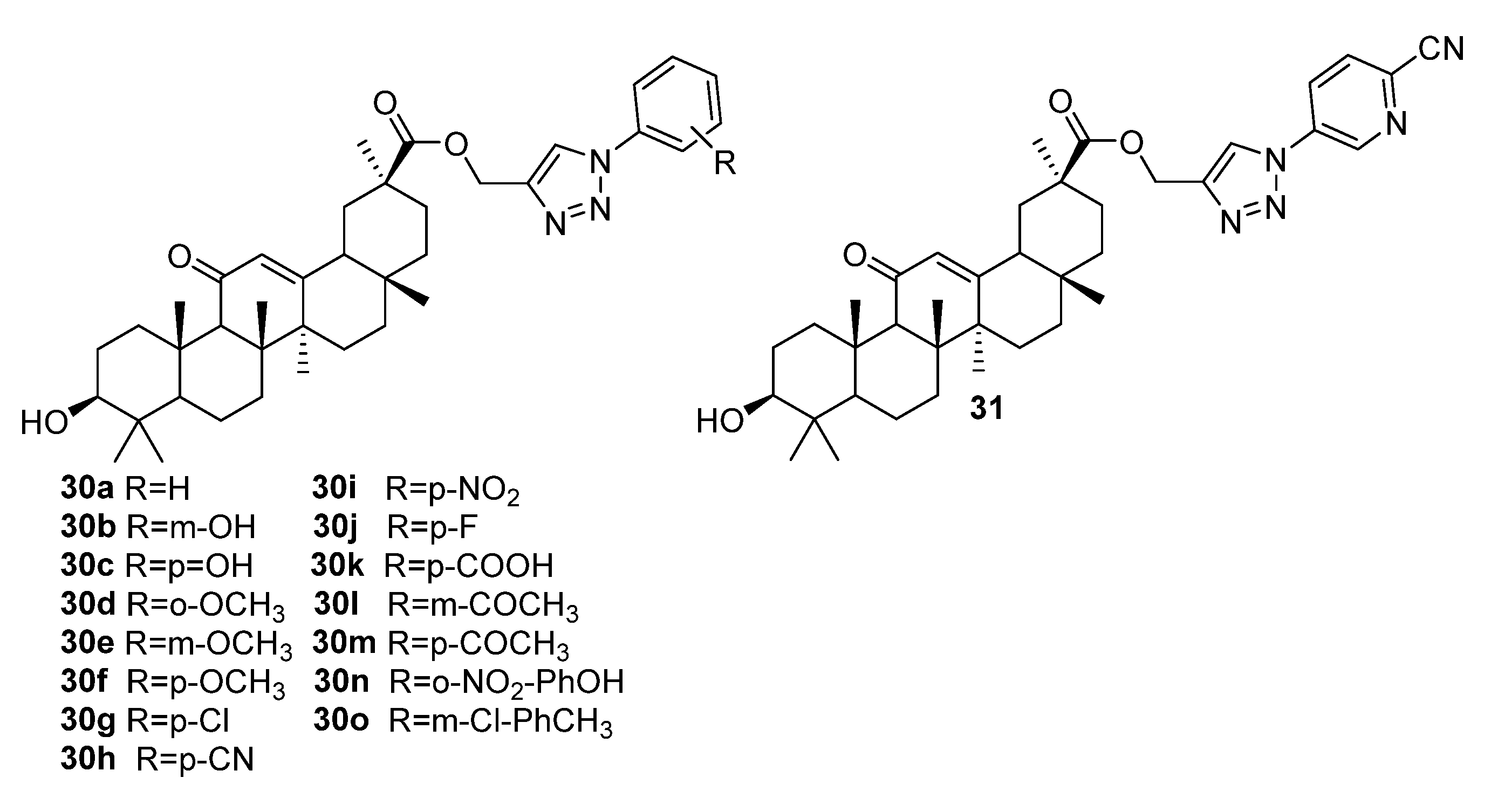













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Parametrs of Activity and Mechanism of Action | Ref. |
|---|---|---|
| Antidiabetic activity | ||
| 22e | PTP1B enzyme inhibitory activity IC50 4.15 μM | [19] |
| 34f | A-glucosidase inhibitory activity IC50 0.22 µM; Ki 0.84 µM Exhibited a competitive type of inhibition: the inhibitor binds with the active site residue of the enzyme. The molecular docking interactions indicated that all compounds were well fitted in the active site of α-glucosidase, where His280, Gln279, Asp215, His351, Arg442, and Arg315 mainly stabilize the binding of these compounds. | [25] |
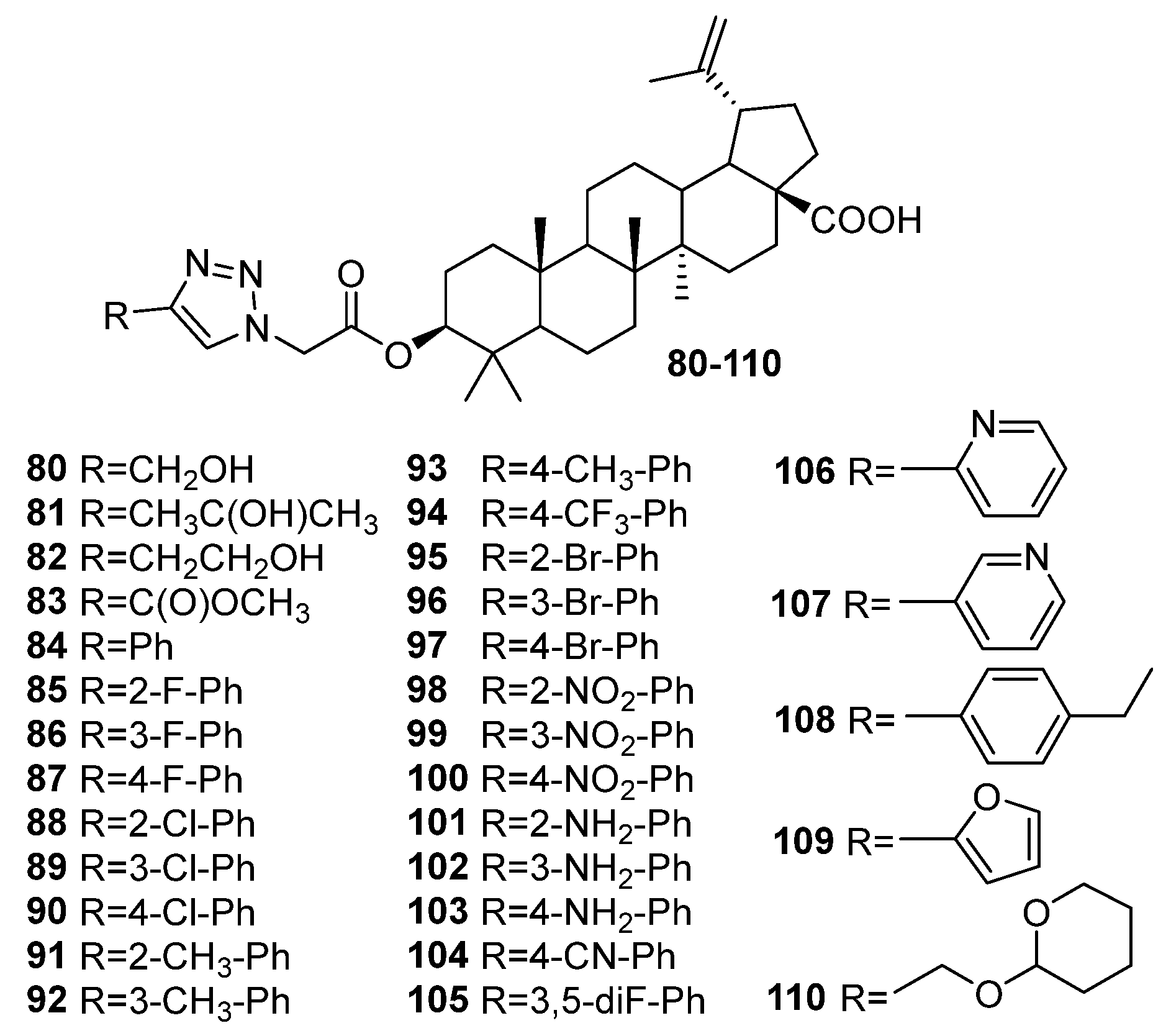
| 103 | A-glucosidase inhibitory activity IC50 2.83 ± 0.19 μM A mixed type inhibitor, interacts with α-glucosidase by changing the secondary structure of the protein, could bind to the active site of α-glucosidase (molecular docking studies), could reduce fasting blood glucose levels in mice and improve glucose tolerance and reverse dyslipidemia (in vivo). | [39] |
| Neuroprotective activity | ||
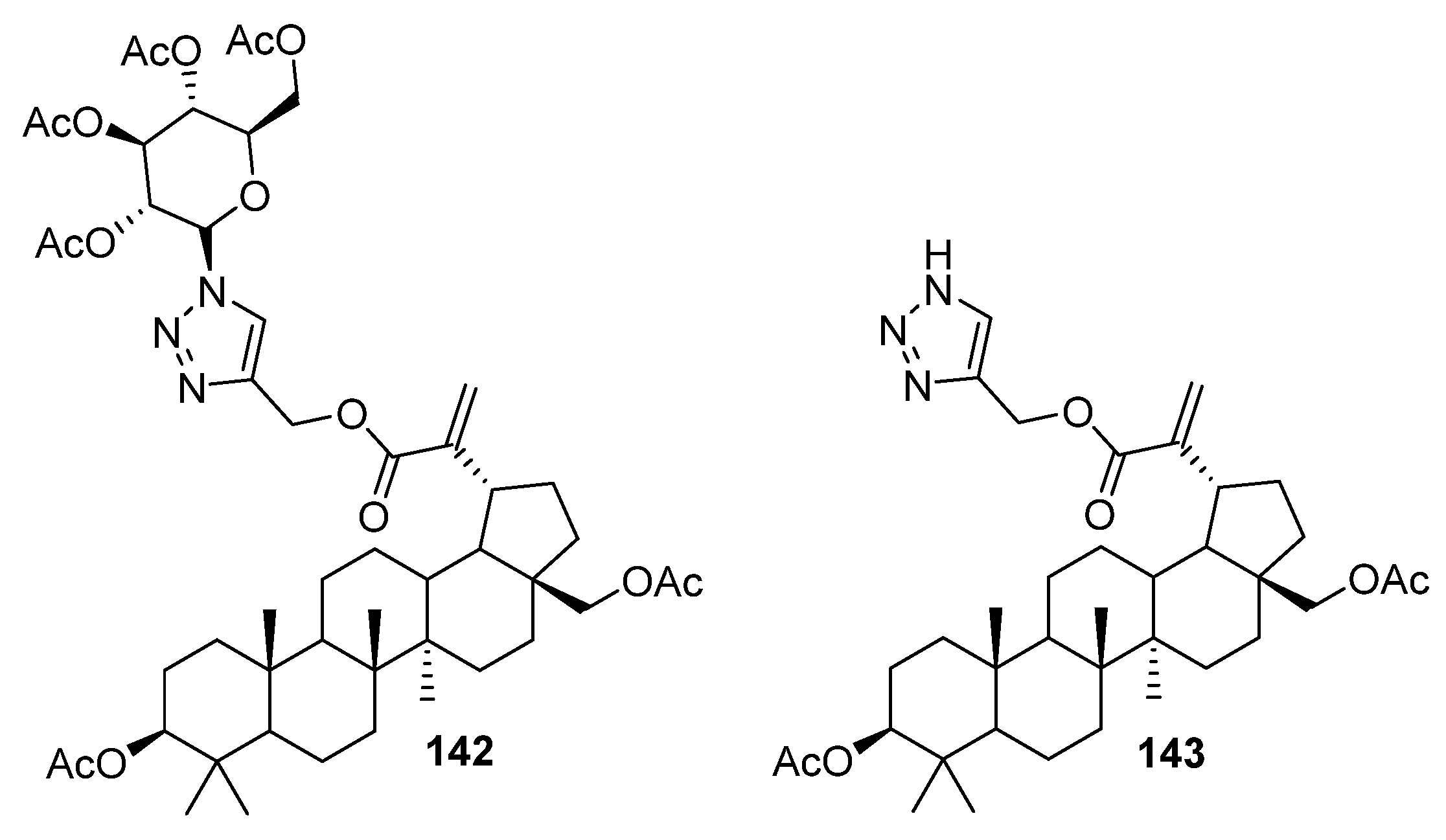
| 142 | Modulated oxidative stress and caspase-3,7 activity, blocked mitochondrial permeability transition pore opening, demonstrated potent restoration of the mitochondrial membrane potential. | [52] |
| Antiviral activity | ||
| 2 | Anti-RSV activity EC50 0.053 µM May act similarly to inosine monophosphate dehydrogenase inhibitors and the active metabolite of ribavirin, leading to guanosine triphosphate depletion. | [13] |
| 36a | Anti-influenza activity against A/WSN/33 (H1N1) IC50 5.20 µM Inhibited viral replication by directly targeting the influenza hemagglutinin protein (KD 1.50 μM), thus efficiently preventing the attachment of the virion to its receptors on host cells and subsequent infection. | [26,27] |
| 43, 45 | Anti-influenza activity against A/WSN/33 (H1N1) 100% inhibition | [28] |
| 50a | Anti-influenza activity against A/WSN/33 (H1N1) IC50 2.86 μM Inhibited influenza A/WSN/33 (H1N1) virus-induced agglutination of cRBCs (red blood cells) in a dose-dependent manner at concentrations of 1.3 μM or more. Similar results were observed for the anti-hemagglutinin antibody at concentrations of 0.063 ng/mL or more. | [29] |
| 65a | Anti-influenza activity against A/WSN/33 (H1N1) IC50 5.47 µM Hemagglutination inhibition assay and docking simulation indicated that the antiviral activity was potentially due to its high affinity to HA protein, thus blocking the attachment of influenza viruses to host cells. | [33] |
| 67a | Anti-influenza activity against A/WSN/33 (H1N1) IC50 5.23 μM Could disrupt the interaction between protein hemagglutinin and its sialic acid receptor, thus blocking virus and host cell recognition. | [34] |
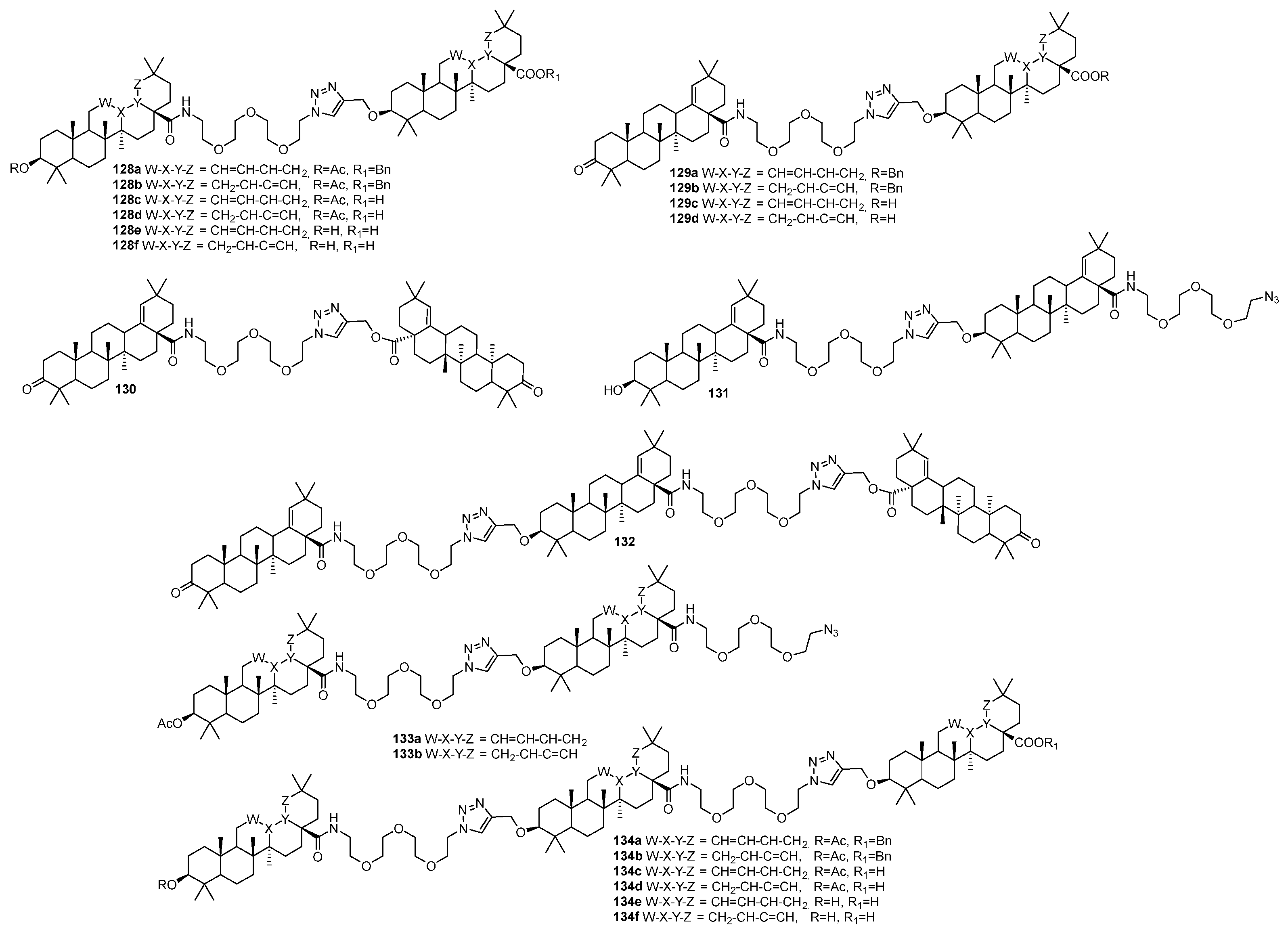
| 128e | Anti-HIV-1 EC50 50.6 µM | [50] |
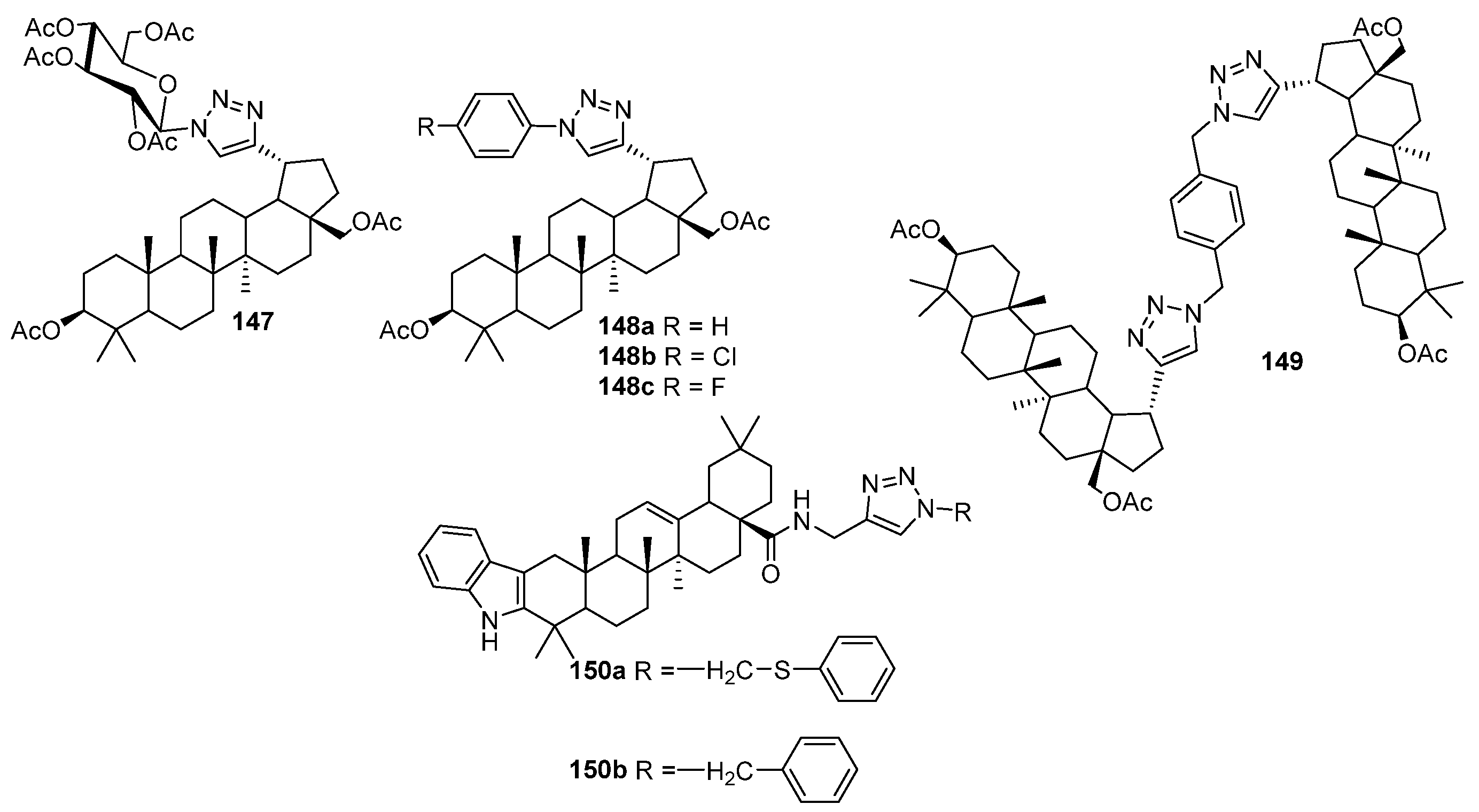
| 148a, 148c | Antiviral activity against human papillomavirus-11 R = H: EC50 < 2.97 µM, SI > 125 R = F: EC50 1.20 µM, SI > 7 | [60] |
| 150b | Antiviral activity against human cytomegalovirus EC50 < 0.05 µM, SI 81 | [60] |
| Antiparasitic, antimicrobial, and antifungal activities | ||
| 6b | Growth-stimulating activity against strains in the range of 20–200% | [15] |
| 25d | Antiparasitic activity against T. gondii IC50 128.0 µM Possessed a strong binding affinity for T. gondii calcium-dependent protein kinase 1. | [20] |
| 30n | Antiplasmodial activity against chloroquine-sensitive (NF-54) strain P. falciparum. IC50 0.47 µM (in vivo) Increased oxidative stress and decreased the metabolic activity of P. falciparum in a dose-dependent manner, induced apoptosis-like cell death (the loss of mitochondrial membrane potential, activation of caspase proteases, and DNA damage). | [23] |
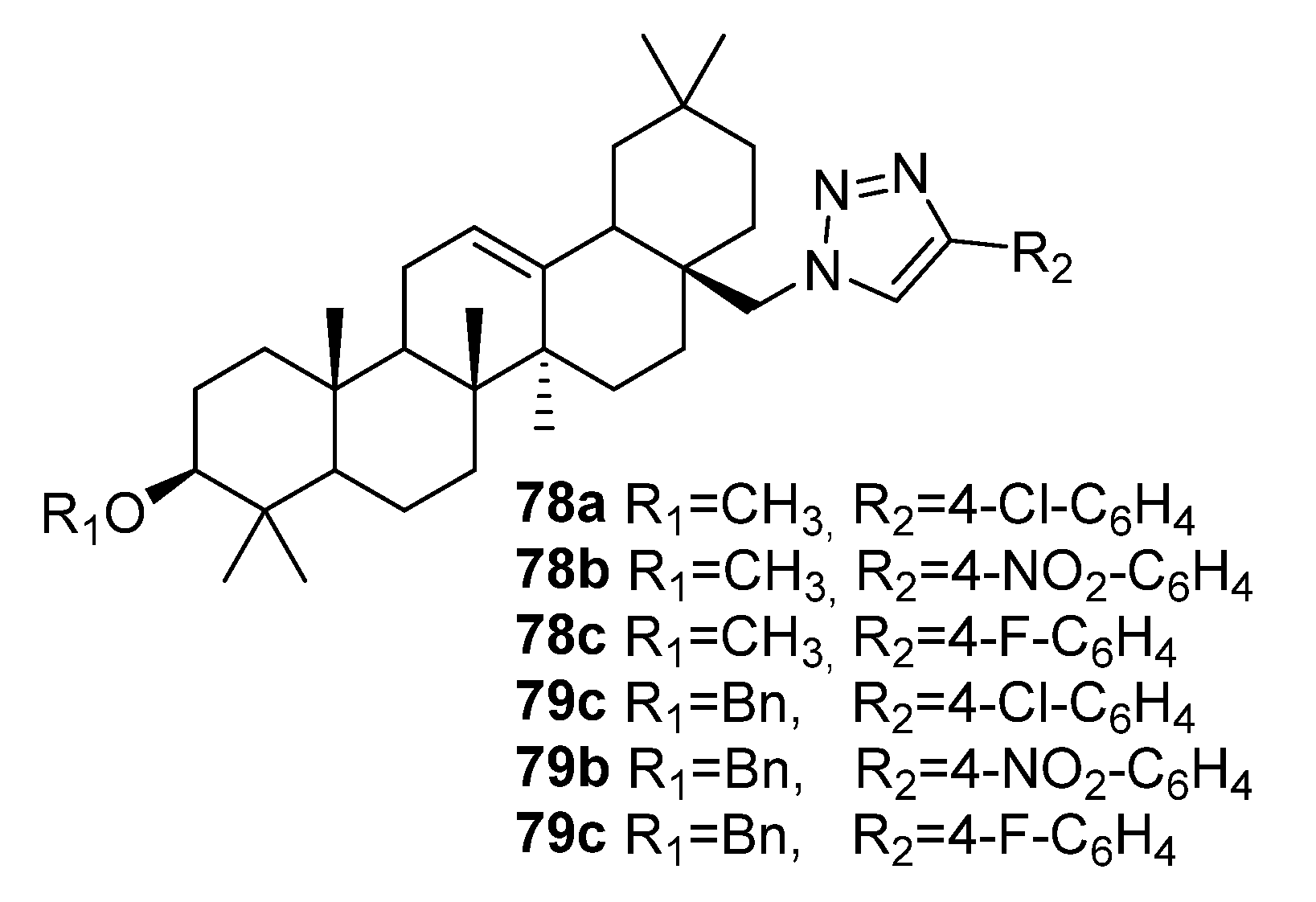
| 79c | Antifungal activity against S. sclerotiorum 89.6% inhibition | [39] |
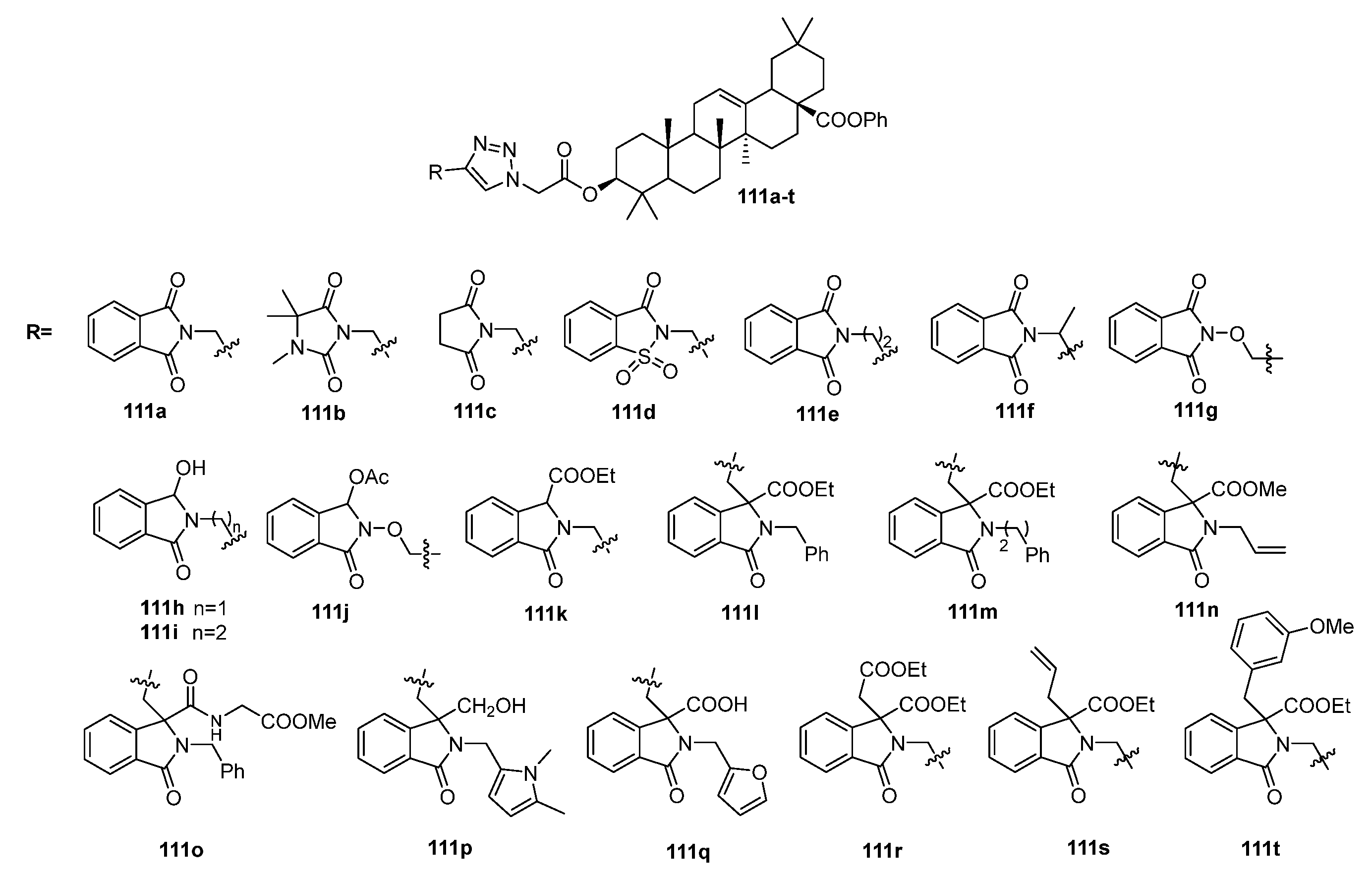
| 111g | Antibacterial activity against L. monocytogenes MIC50 9.48 µM The in silico molecular docking study revealed that the compound can fit well into the binding cavity of the ABC substrate-binding protein Lmo0181 from L. monocytogenes. | [41] |
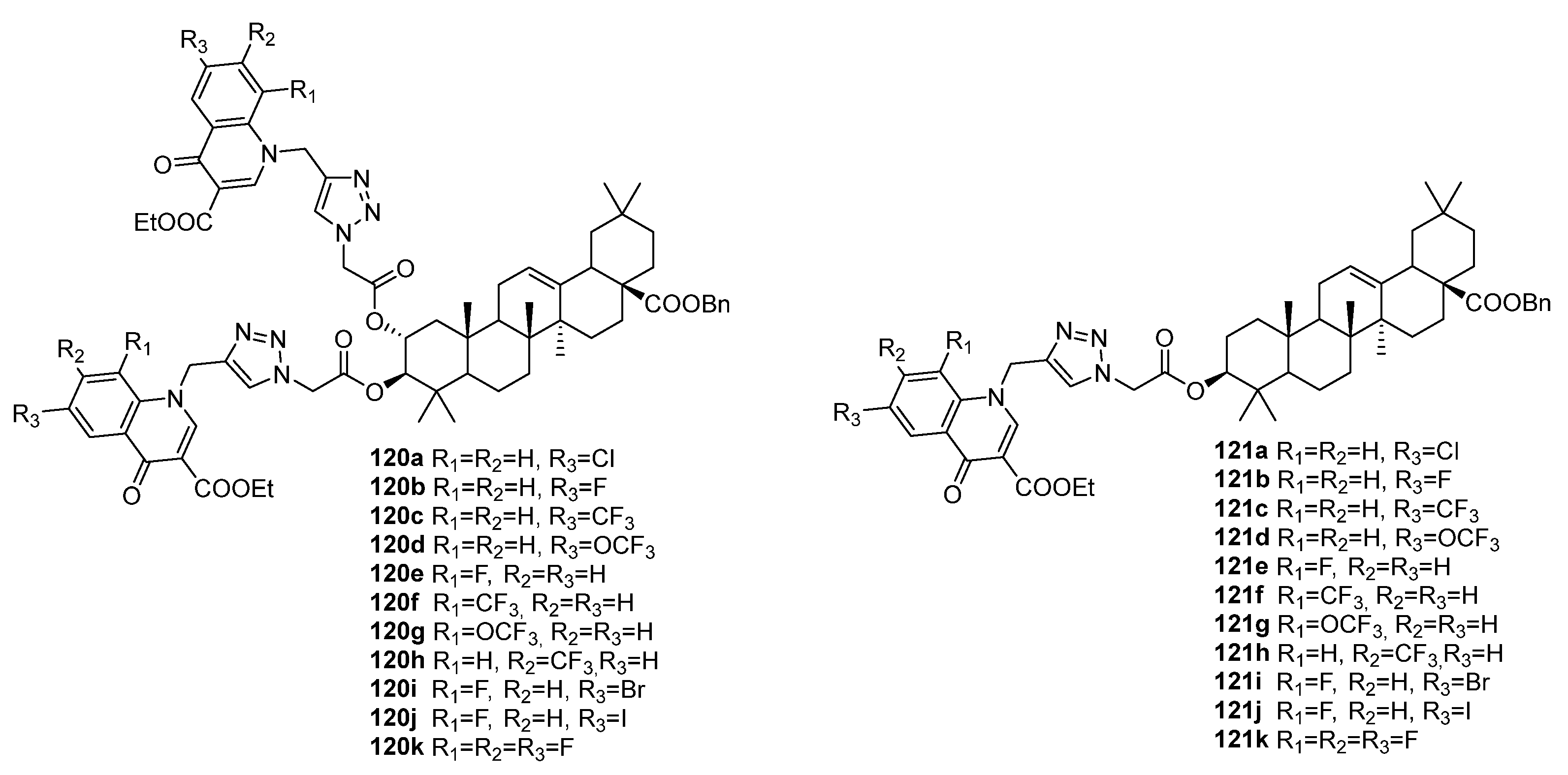
| 120c | Antibacterial activity against E. faecalis, E. coli, and P. aeruginosa MIC 3.25 µg/mL (for all strains) | [45] |
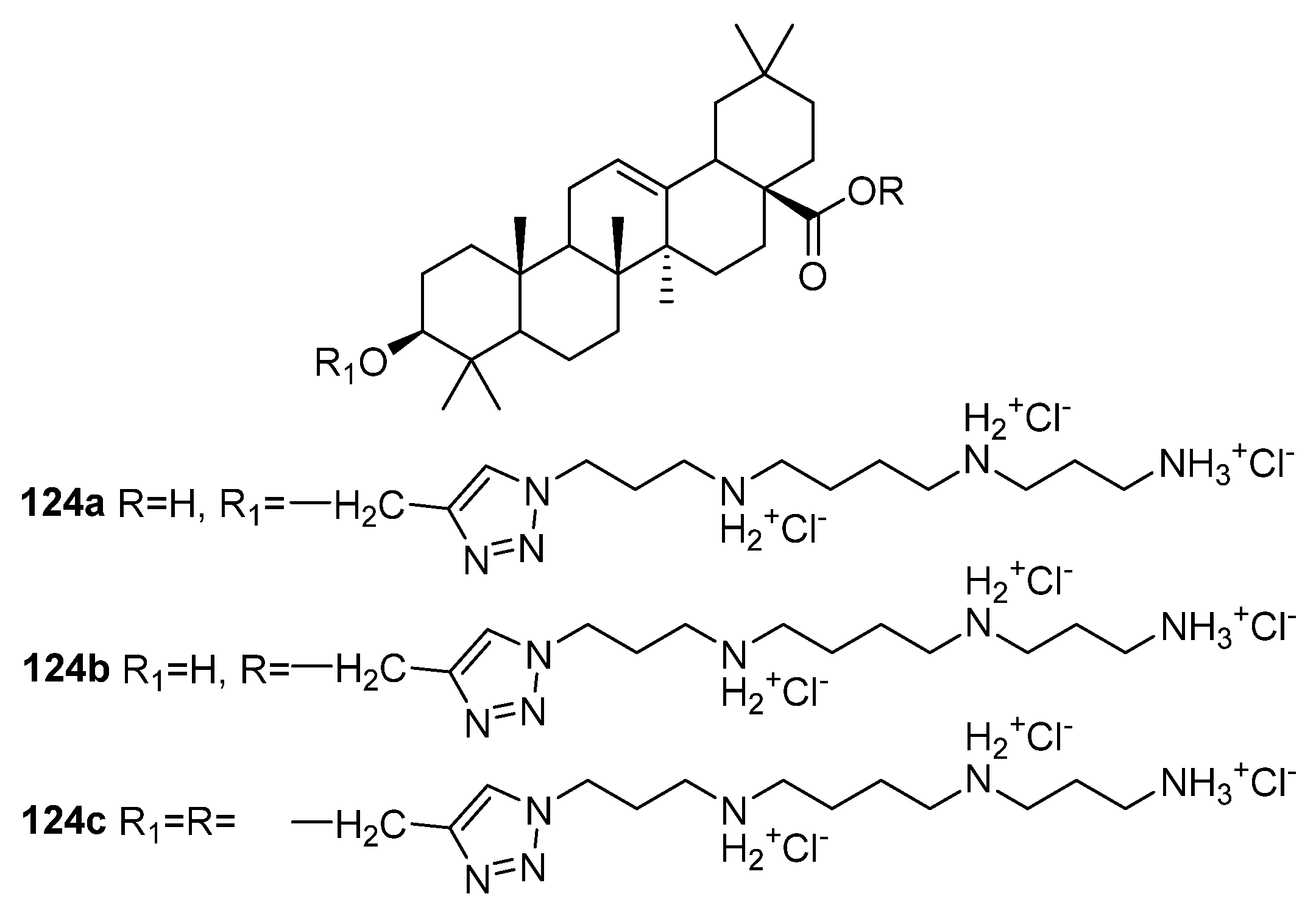
| 124a | Antimicrobial activity against L. acidophilus IC50 < 1.13 µM Proposed mechanism of action is based on the disturbing of the membrane integrity of bacteria by interaction with negative charges of phosphate groups of the LPS on the surface of the outer membrane. | [47,48] |
| 128e | Antimicrobial inhibitory activity against S. aureus and E. coli >50% (c 25 µg/mL) | [50] |
| 134f | Antimicrobial inhibitory activity against S. aureus >40% (c 25 µg/mL) | [50] |
| 130 | Antimicrobial inhibitory activity against E. faecalis >62% (c 25 µg/mL) | [50] |
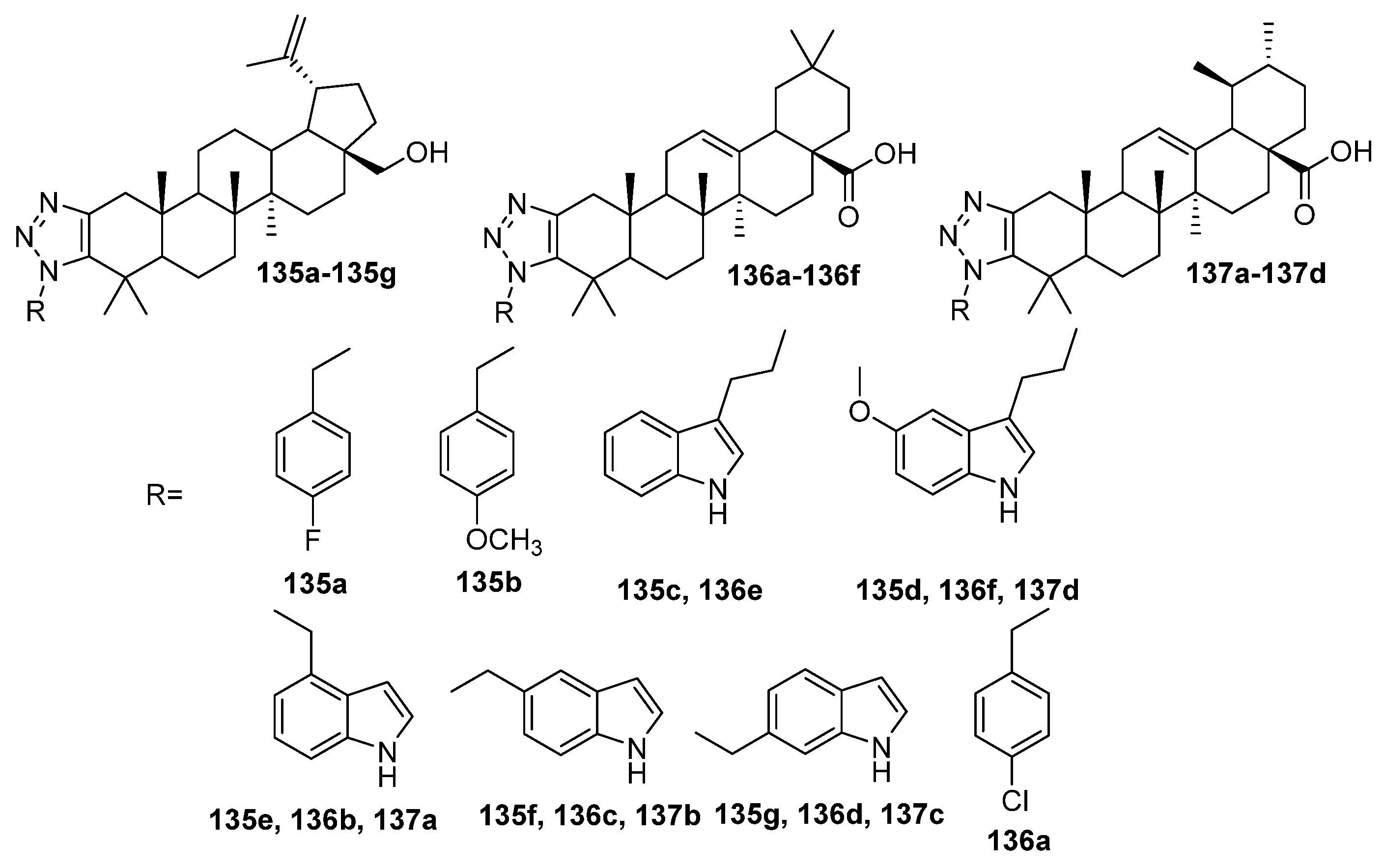
| 135c | Antimicrobial activity against S. enterica subsp. enterica MIC50 16.61 mg/mL | [51] |
| Anti-inflammatory activity | ||
| 22b | Anti-inflammatory activity COX-2 inhibition IC50 1.16 µM, SI 64.66 | [21] |
| 33b | Suppressed the expression of pro-inflammatory cytokines including IL-6, TNF-α and NO, it also suppressed the expression of iNOS and COX-2 in LPS-induced RAW264.7 cells in a dose-dependent manner. Western blot results indicated that the suppressing effect on pro-inflammatory cytokines was correlated with the suppression of NF-κB and MAPK signaling pathways. | [24] |
| Anticancer activity | ||
| 13b | Cytotoxicity against breast cancer cell line MCF-7 IC50 1.55 µM Molecular docking data supported by flow cytometry analysis showed that the most likely mechanism of cytotoxic activity was the affinity to Mdm2 binding sites. | [18] |
| 10a | Cytotoxicity against breast cancer cell line T47D IC50 1.20 µM Activated the mitochondrial apoptosis pathway in A549 cells. The molecular docking study showed that the interaction of the 1,4-quinone moiety with the enzyme active site through hydrogen bond led to an increase in activity. | [16] |
| 29 | Anticancer activity against A549 (IC50 1.89 µM), Huh7 (IC50 1.87 µM), and HepG2 (IC50 1.77 µM) cell lines. Induced significant degradation of MDM2 (only 25% to that of SM1) and promoted the expression of P21 and PUMA proteins, thus inhibiting the proliferation (77.67% of 1B vs. 60.37% of CON in G1 phase) and promoting the apoptosis (26.74% of 1B vs. 3.35% of CON) of A549 cells. | [22] |
| 60c | Cytotoxicity against all cancer cell lines IC50 range 0.60–1.60 µM Apoptosis via the mitochondrial pathway, inhibited growth and disintegrated spheroid cultures of HCT116 and HeLa cells, which would be important for the treatment of solid tumors. | [31] |
| 68, 69 | Antiproliferative activity against ovarian cancer cell line A2780 IC50 5.9 µM for 68; IC50 9.4 µM for 69 Could inhibit cell metastasis and invasion through damage of actin dynamics and downregulation of MMP2/MMP9 proteins. Mitochondrial accumulation of metal-arene complexes caused mitochondrial membrane potential damage, oxidative phosphorylation, ATP depletion, and autophagy. Displayed excellent activity to disintegrate 3D multicellular tumor spheroids, showing potential for the treatment of solid tumors. | [35] |
| 71k | Cytotoxicity against lung cancer cell line A549 IC50 2.67 µM Inhibited NF-κB DNA binding, the activity of NF-κB, nuclear translocation, and IκBα phosphorylation. The treatment of A549 cells induced apoptosis and inhibited in vitro cell migration. May be a potential NF-κB inhibitor with the ability to induce apoptosis and suppress cell migration. | [36] |
| 73i | Cytotoxicity against glioma cell line U251 IC50 0.94 µM Induced necroptosis mainly by activating the RIP1/RIP3/MLKL pathway, exerted acceptable BBB permeability in mice, and inhibited U251 cell proliferation in an in vivo zebrafish xenograft model. | [37] |
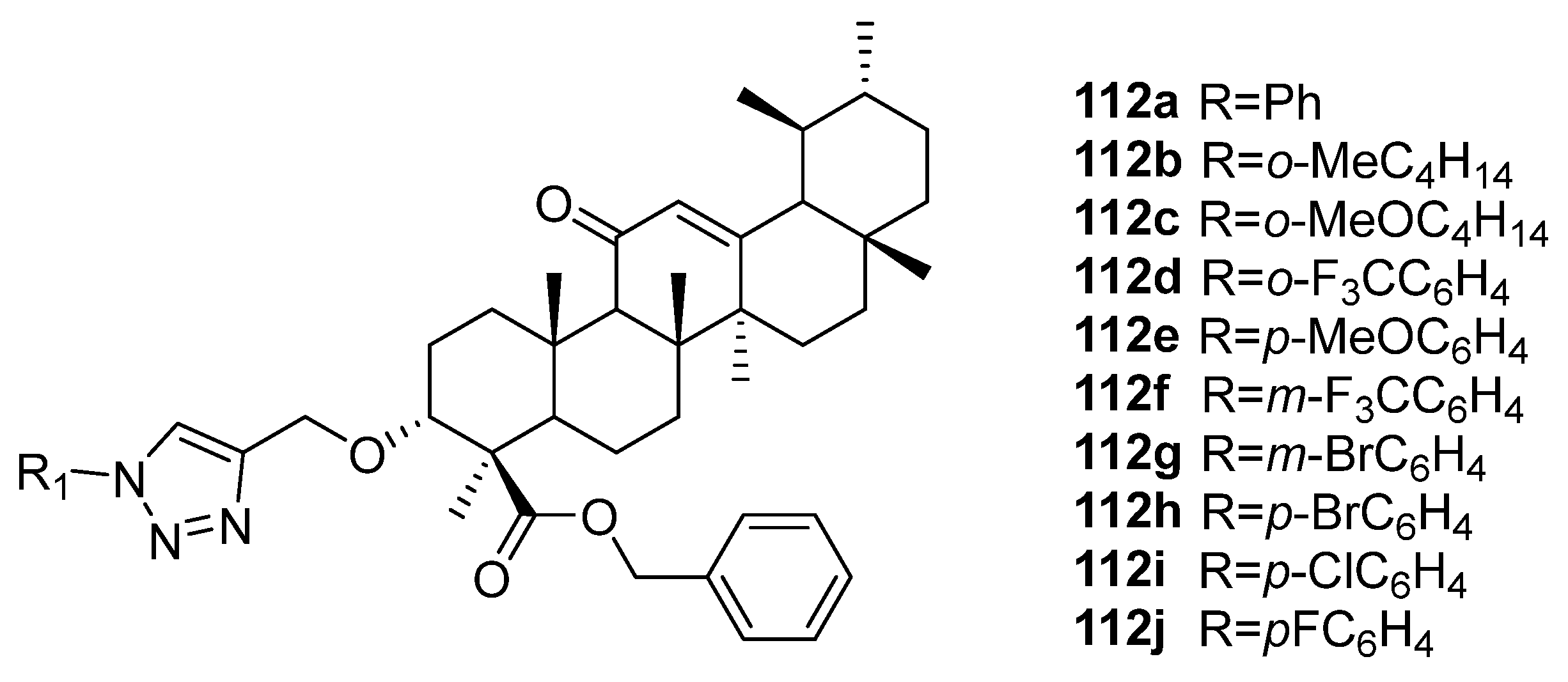
| 112j | Cytotoxicity against breast cancer cell line MDA-MB-231 IC50 4.45 µM Can target CHK1 to produce anticancer effects in TNBC (triple negative breast cancer). | [42] |
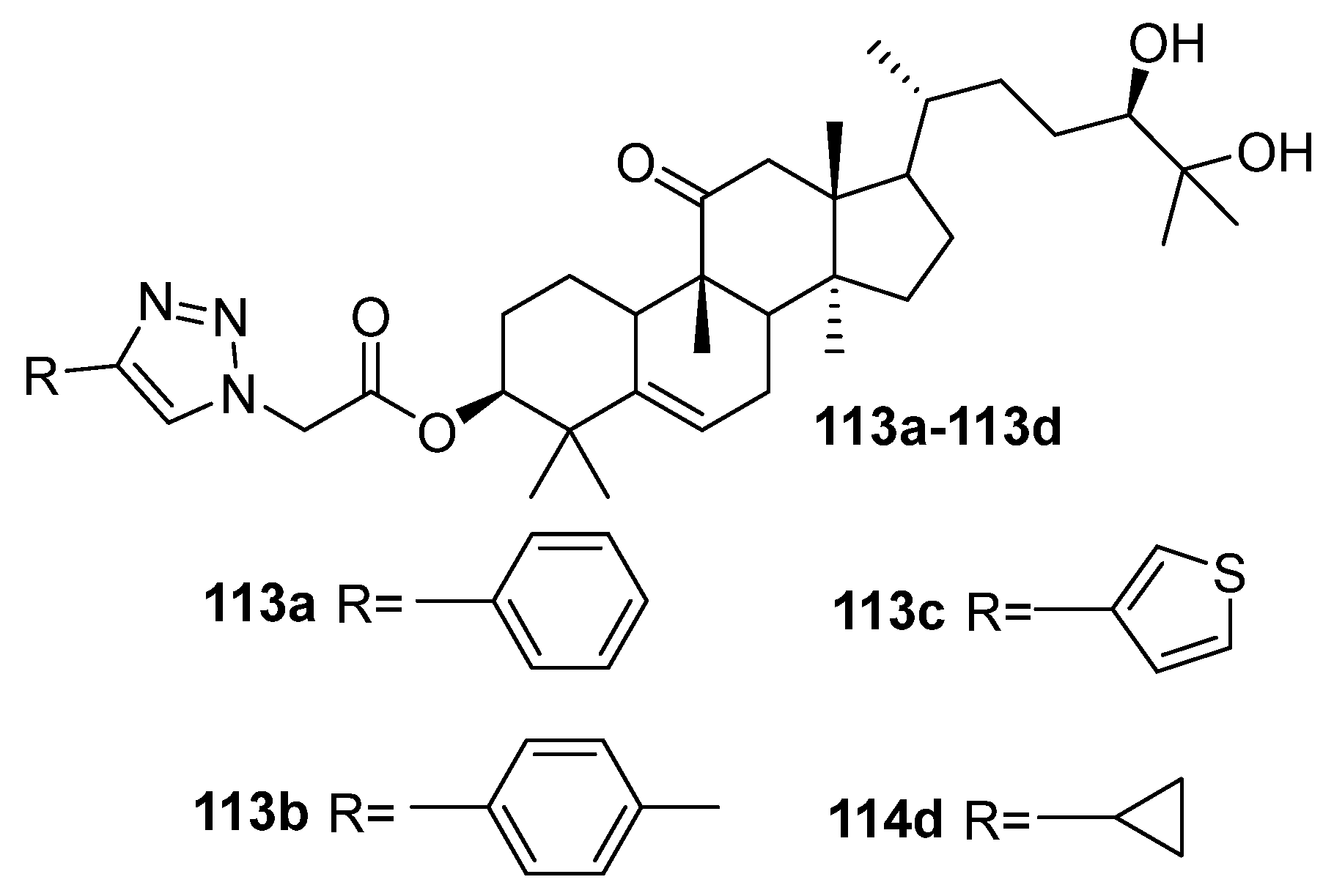
| 113a | Cytotoxicity against lung cancer cell line A549 IC50 7.19 µM Induced G1 phase arrest in A549 cell lines, regulated the signal transducer and activator of transcription 3 signal pathway by inhibiting phosphorylation of JanusKinase 2 and STAT3 and simultaneously increasing the protein level of downstream cyclin p21. | [43] |
| 123a | Cytotoxicity against human T-lymphoblastic leukemia cancer cell line IC50 6.5 µM | [46] |
| 125b | Cytotoxicity against breast cancer cell line MCF-7 IC50 3.0 µM | [49] |
| 129d | Cytotoxicity against melanoma cancer cell line G-361 IC50 20.0 µM | [50] |
| 136f | Cytotoxicity against leukemia cell line HL-60 cells IC50 1.3 µM Caused mitochondrial dysfunction and arrested the cell cycle in the G0/G1 phase to induce apoptosis of HL-60 cells, also induced autophagy and inhibited the proliferation of HL-60 cells. | [51] |
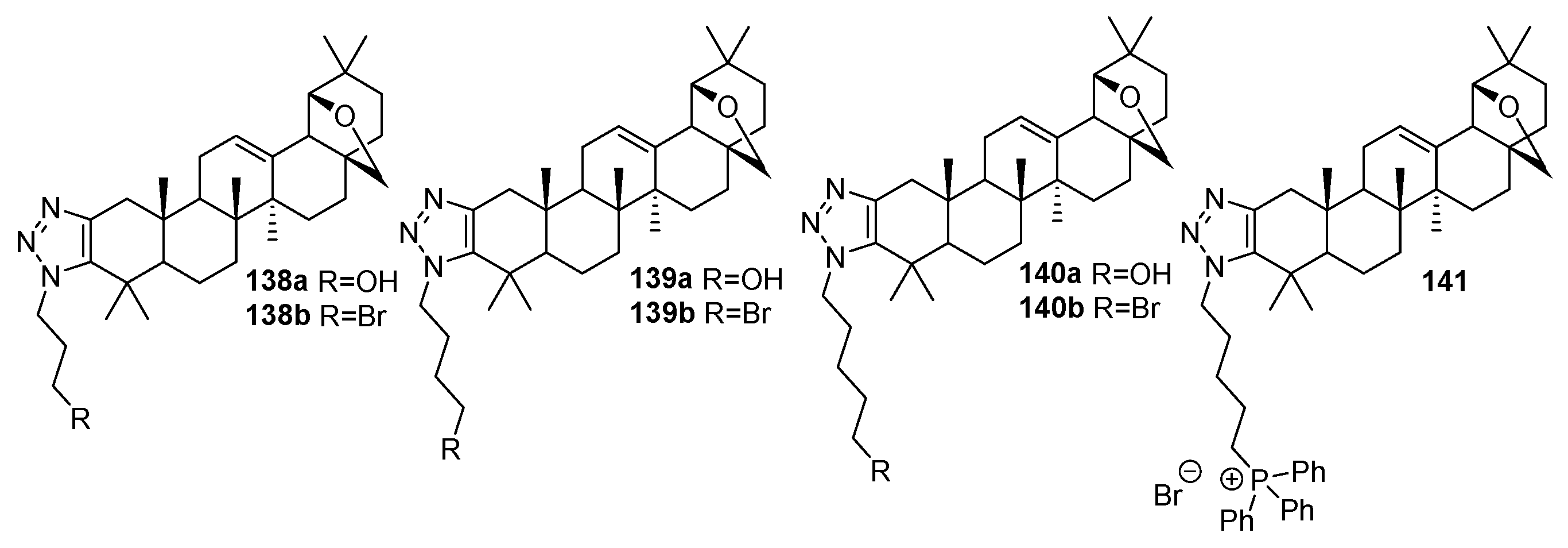
| 141 | Cytotoxicity against Hela (IC50 1.19 µM), Hep92 (IC50 1.15 µM), MCF-7 (IC50 1.14 µM), MDA-MB-231 (IC50 1.12 µM), and SGC-7901 (IC50 1.05 µM) cells Could induce SGC-7901 cell apoptosis through the mitochondrial pathway, inducing cell cycle arrest. Could reduce the expression of Vimentin and increase the expression of E-cadherin to hinder the epithelial–esenchymal transition. | [52] |
| 144a | Cytotoxicity against melanoma cancer cell line RPMI 88.3% inhibition at a dose of 2 µM May induce apoptosis by disrupting the Bcl2/Bax ratio. | [54] |
| 144b | Cytotoxicity against melanoma cancer cell line RPMI-7951 IC50 18.8 µM Revealed a mitochondria-level induced apoptotic mechanism, inhibited mitochondrial respiration in RPMI−7951 cells, caused a significant decrease of anti-apoptotic Bcl-2 gene expression, while increasing pro-apoptotic BAX gene expression. | [55] |
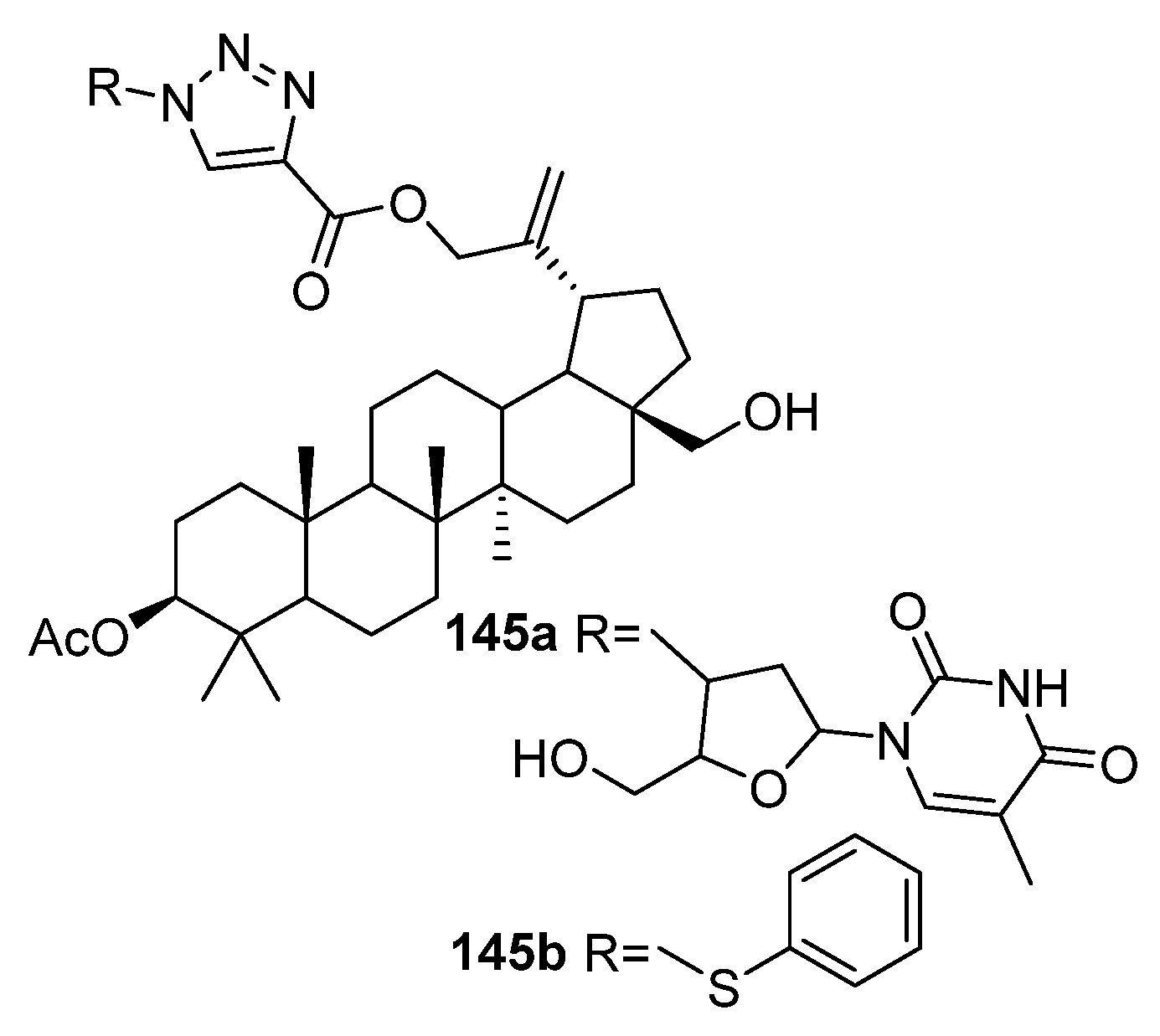
| 145a | Cytotoxicity against breast cancer cell line MCF-7 IC50 4.37 µM | [56] |
| 152d | Antiproliferative activity against human cancer cell lines SMMC-7721 (IC50 5.57 µM), HepG2 (IC50 7.49 µM), MNK-45 (IC50 6.31 µM), SW620 (IC50 6.00 µM), and A549 (IC50 5.79 µM) Induced cell apoptosis and autophagy in SMMC cells, resulting in antiproliferation and G0/G1 cell cycle arrest by regulating protein expression levels of Bax, Bcl-2, and LC3. | [61] |
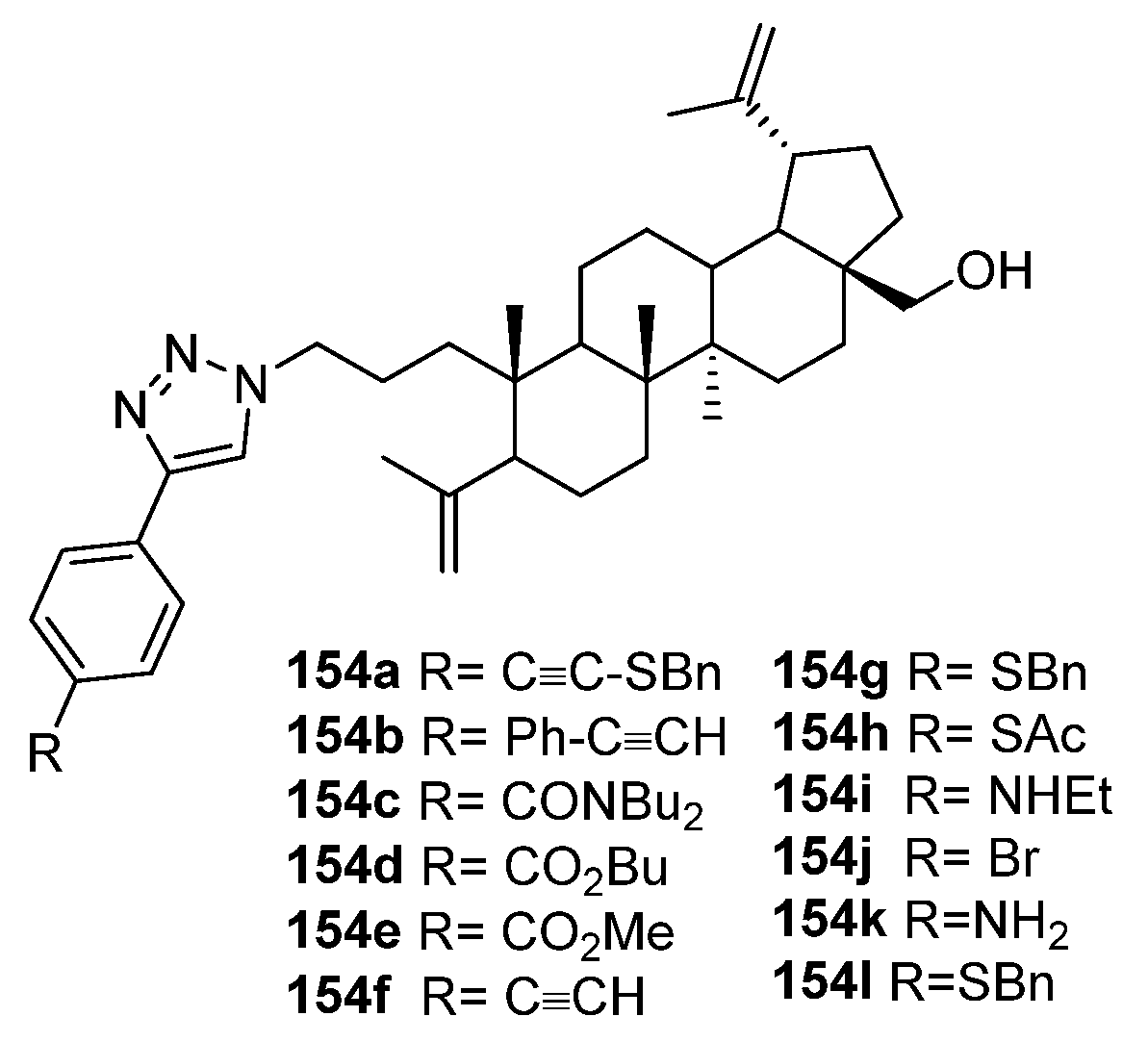
| 154l | Cytotoxicity against breast cancer cell line MCF-7 IC50 6.5 µM | [63] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrova, A.V.; Kazakova, O.B. 1,3-Dipolar Cycloaddition and Mannich Reactions of Alkynyl Triterpenes: New Trends in Synthetic Strategies and Pharmacological Applications. Int. J. Mol. Sci. 2025, 26, 4329. https://doi.org/10.3390/ijms26094329
Petrova AV, Kazakova OB. 1,3-Dipolar Cycloaddition and Mannich Reactions of Alkynyl Triterpenes: New Trends in Synthetic Strategies and Pharmacological Applications. International Journal of Molecular Sciences. 2025; 26(9):4329. https://doi.org/10.3390/ijms26094329
Chicago/Turabian StylePetrova, Anastasiya V., and Oxana B. Kazakova. 2025. "1,3-Dipolar Cycloaddition and Mannich Reactions of Alkynyl Triterpenes: New Trends in Synthetic Strategies and Pharmacological Applications" International Journal of Molecular Sciences 26, no. 9: 4329. https://doi.org/10.3390/ijms26094329
APA StylePetrova, A. V., & Kazakova, O. B. (2025). 1,3-Dipolar Cycloaddition and Mannich Reactions of Alkynyl Triterpenes: New Trends in Synthetic Strategies and Pharmacological Applications. International Journal of Molecular Sciences, 26(9), 4329. https://doi.org/10.3390/ijms26094329