
Targeting Gene C9orf72 Pathogenesis for Amyotrophic Lateral Sclerosis

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
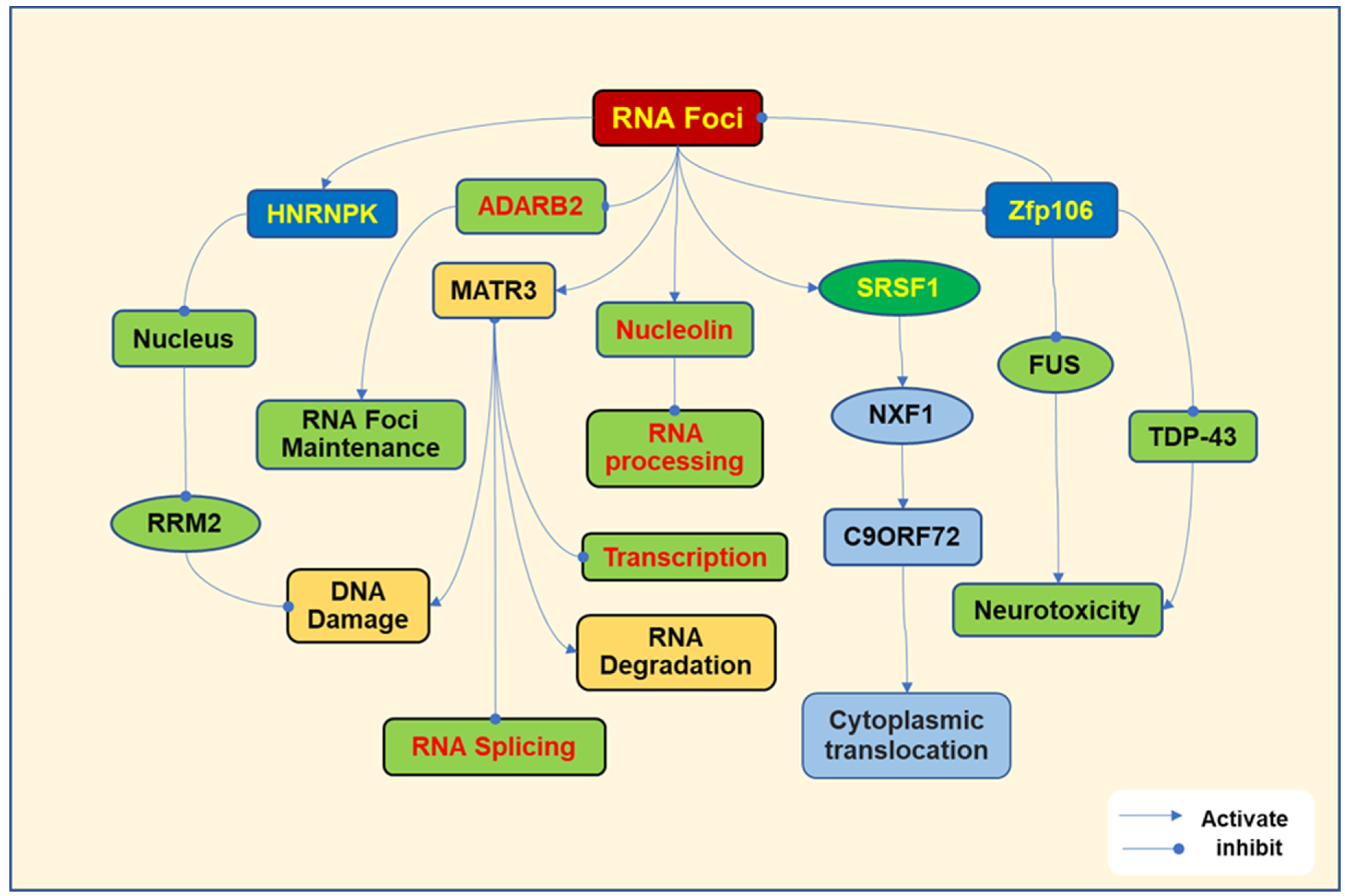
2. Pathogenesis Due to Toxic RNA Foci
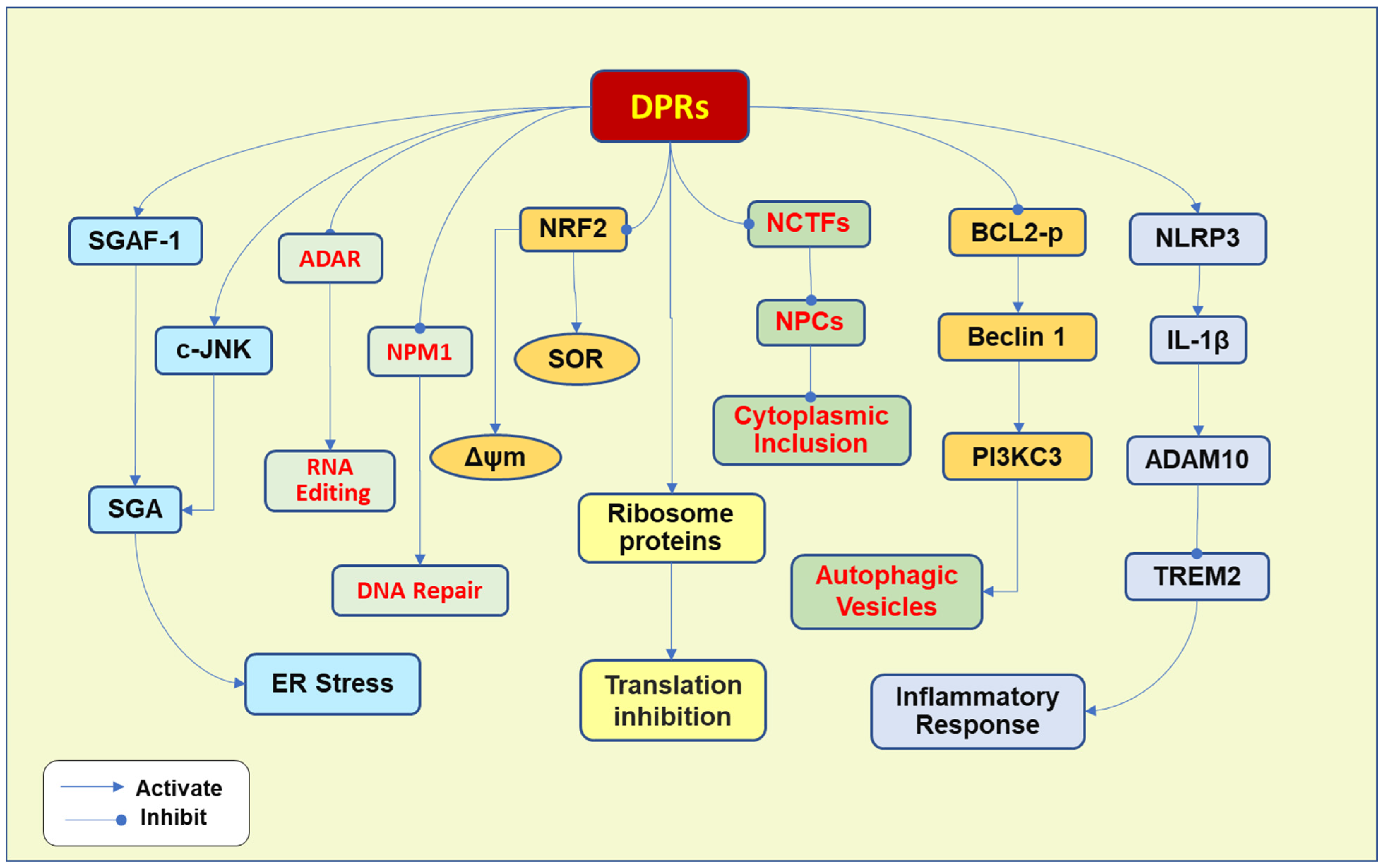
3. Pathogenesis Due to Toxic DPRs
3.1. Translational Inhibition and Nucleic Acid Repair
3.2. Induction of Oxidative Stress
3.3. Inhibition of Ubiquitin-Proteasome and Autophagy
3.4. Induction of Inflammation
3.5. Impairment of Nucleocytoplasmic Transport (NCT)
3.6. The Dysfunction of Synapses
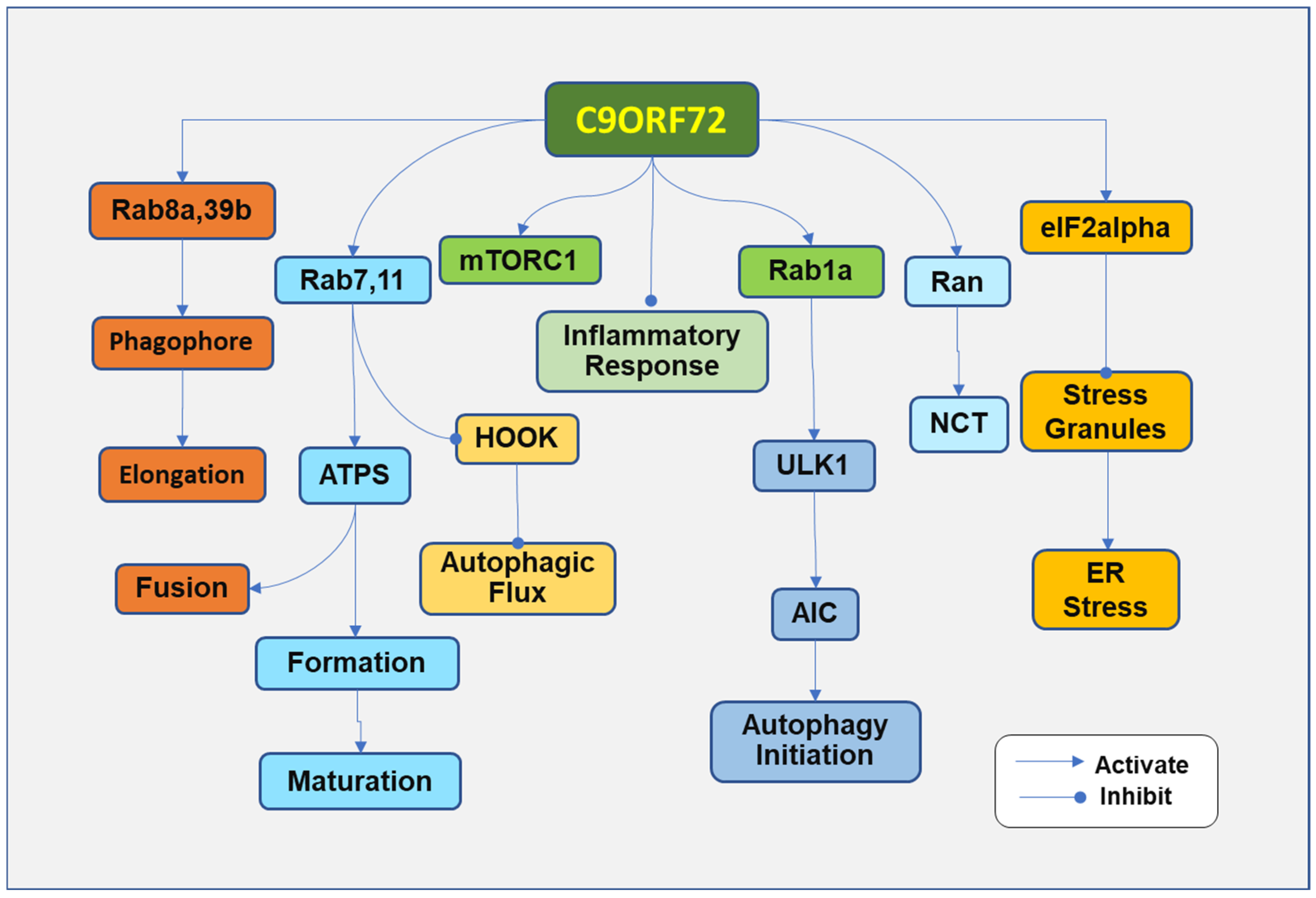
4. Pathogenesis Due to the Loss of C9ORF7 Function
4.1. Impairment of Autophagy
4.2. Dysfunction of NCT
4.3. ER Stress
4.4. Inflammation
5. Targeting C9orf722 HRE for Potential Therapeutic Strategies
5.1. Targeting RNA Repeats and DRPs
5.1.1. ASOs
5.1.2. Gene Editing (CRISPR/Cas9)
5.1.3. Small Molecules
5.2. Targeting Oxidative Stress
5.3. Autophagic Modulators
5.4. Targeting Neuromuscular Junction (NMJ)
5.5. Targeting NCT
5.6. Targeting Inflammation
6. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Feldman, E.L.; Goutman, S.A.; Petri, S.; Mazzini, L.; Savelieff, M.G.; Shaw, P.J.; Sobue, G. Amyotrophic lateral sclerosis. Lancet 2022, 400, 1363–1380. [Google Scholar] [CrossRef]
- Roggenbuck, J. C9orf72 and the Care of the Patient with ALS or FTD: Progress and Recommendations After 10 Years. Neurol. Genet. 2021, 7, e542. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.; Bakulski, K.; Guo, K.; Hur, J.; Zhao, L.; Saez-Atienzar, S.; Stark, A.; Chia, R.; Garcia-Redondo, A.; Rojas-Garcia, R.; et al. Cumulative Genetic Score and C9orf72 Repeat Status Independently Contribute to Amyotrophic Lateral Sclerosis Risk in 2 Case-Control Studies. Neurol. Genet. 2023, 9, e200079. [Google Scholar] [CrossRef]
- Simon-Sanchez, J.; Dopper, E.G.; Cohn-Hokke, P.E.; Hukema, R.K.; Nicolaou, N.; Seelaar, H.; de Graaf, J.R.; de Koning, I.; van Schoor, N.M.; Deeg, D.J.; et al. The clinical and pathological phenotype of C9ORF72 hexanucleotide repeat expansions. Brain 2012, 135, 723–735. [Google Scholar] [CrossRef]
- Gijselinck, I.; Van Langenhove, T.; van der Zee, J.; Sleegers, K.; Philtjens, S.; Kleinberger, G.; Janssens, J.; Bettens, K.; Van Cauwenberghe, C.; Pereson, S.; et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: A gene identification study. Lancet Neurol. 2012, 11, 54–65. [Google Scholar] [CrossRef]
- Smeyers, J.; Banchi, E.G.; Latouche, M. C9ORF72: What It Is, What It Does, and Why It Matters. Front. Cell Neurosci. 2021, 15, 661447. [Google Scholar] [CrossRef]
- Rizzu, P.; Blauwendraat, C.; Heetveld, S.; Lynes, E.M.; Castillo-Lizardo, M.; Dhingra, A.; Pyz, E.; Hobert, M.; Synofzik, M.; Simon-Sanchez, J.; et al. C9orf72 is differentially expressed in the central nervous system and myeloid cells and consistently reduced in C9orf72, MAPT and GRN mutation carriers. Acta Neuropathol. Commun. 2016, 4, 37. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Heckman, M.G.; Dejesus-Hernandez, M.; Baker, M.C.; Soto-Ortolaza, A.I.; Rayaprolu, S.; Stewart, H.; Finger, E.; Volkening, K.; Seeley, W.W.; et al. Length of normal alleles of C9ORF72 GGGGCC repeat do not influence disease phenotype. Neurobiol. Aging 2012, 33, 2950.e5–2950.e7. [Google Scholar] [CrossRef]
- Gami, P.; Murray, C.; Schottlaender, L.; Bettencourt, C.; De Pablo Fernandez, E.; Mudanohwo, E.; Mizielinska, S.; Polke, J.M.; Holton, J.L.; Isaacs, A.M.; et al. A 30-unit hexanucleotide repeat expansion in C9orf72 induces pathological lesions with dipeptide-repeat proteins and RNA foci, but not TDP-43 inclusions and clinical disease. Acta Neuropathol. 2015, 130, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Li, Y.; Li, Y. Meta-analysis of the association between C9orf72 repeats and neurodegeneration diseases. J. Neurogenet. 2024, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gijselinck, I.; Van Mossevelde, S.; van der Zee, J.; Sieben, A.; Engelborghs, S.; De Bleecker, J.; Ivanoiu, A.; Deryck, O.; Edbauer, D.; Zhang, M.; et al. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol. Psychiatry 2016, 21, 1112–1124. [Google Scholar] [CrossRef]
- Nagy, Z.F.; Pal, M.; Engelhardt, J.I.; Molnar, M.J.; Klivenyi, P.; Szell, M. Beyond C9orf72: Repeat expansions and copy number variations as risk factors of amyotrophic lateral sclerosis across various populations. BMC Med. Genom. 2024, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lin, Z.; Chen, X.; Cao, B.; Wei, Q.; Ou, R.; Zhao, B.; Song, W.; Wu, Y.; Shang, H.F. Large C9orf72 repeat expansions are seen in Chinese patients with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging. 2016, 38, 217.e15–217.e22. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Jiao, B.; Shen, L. The Development of C9orf72-Related Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Disorders. Front. Genet. 2020, 11, 562758. [Google Scholar] [CrossRef]
- Beckers, J.; Van Damme, P. Toxic gain-of-function mechanisms in C9orf72 ALS-FTD neurons drive autophagy and lysosome dysfunction. Autophagy 2024, 20, 2102–2104. [Google Scholar] [CrossRef]
- Jaroszynska, N.; Salzinger, A.; Tsarouchas, T.M.; Becker, C.G.; Becker, T.; Lyons, D.A.; MacDonald, R.B.; Keatinge, M. C9ORF72 Deficiency Results in Neurodegeneration in the Zebrafish Retina. J. Neurosci. 2024, 44, e2128232024. [Google Scholar] [CrossRef]
- Freibaum, B.D.; Taylor, J.P. The Role of Dipeptide Repeats in C9ORF72-Related ALS-FTD. Front. Mol. Neurosci. 2017, 10, 35. [Google Scholar] [CrossRef]
- Cooper-Knock, J.; Higginbottom, A.; Stopford, M.J.; Highley, J.R.; Ince, P.G.; Wharton, S.B.; Pickering-Brown, S.; Kirby, J.; Hautbergue, G.M.; Shaw, P.J. Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta Neuropathol. 2015, 130, 63–75. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Baskerville, V.; Rapuri, S.; Mehlhop, E.; Jafar-Nejad, P.; Rigo, F.; Bennett, F.; Mizielinska, S.; Isaacs, A.; Coyne, A.N. G(2)C(4) targeting antisense oligonucleotides potently mitigate TDP-43 dysfunction in human C9orf72 ALS/FTD induced pluripotent stem cell derived neurons. Acta Neuropathol. 2023, 147, 1. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.C.; Hsieh, T.C.; Tsai, K.J. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: From pathomechanisms to therapeutic strategies. Ageing Res. Rev. 2024, 100, 102441. [Google Scholar] [CrossRef] [PubMed]
- Braems, E.; Bercier, V.; Van Schoor, E.; Heeren, K.; Beckers, J.; Fumagalli, L.; Dedeene, L.; Moisse, M.; Geudens, I.; Hersmus, N.; et al. HNRNPK alleviates RNA toxicity by counteracting DNA damage in C9orf72 ALS. Acta Neuropathol. 2022, 144, 465–488. [Google Scholar] [CrossRef] [PubMed]
- Maurel, C.; Madji-Hounoum, B.; Thepault, R.A.; Marouillat, S.; Brulard, C.; Danel-Brunaud, V.; Camdessanche, J.P.; Blasco, H.; Corcia, P.; Andres, C.R.; et al. Mutation in the RRM2 domain of TDP-43 in Amyotrophic Lateral Sclerosis with rapid progression associated with ubiquitin positive aggregates in cultured motor neurons. Amyotroph. Lateral Scler. Frontotemporal Degener. 2018, 19, 149–151. [Google Scholar] [CrossRef]
- Sprunger, M.L.; Lee, K.; Sohn, B.S.; Jackrel, M.E. Molecular determinants and modifiers of Matrin-3 toxicity, condensate dynamics, and droplet morphology. iScience 2022, 25, 103900. [Google Scholar] [CrossRef]
- Xu, L.; Li, J.; Tang, L.; Zhang, N.; Fan, D. MATR3 mutation analysis in a Chinese cohort with sporadic amyotrophic lateral sclerosis. Neurobiol. Aging 2016, 38, 218.e3–218.e4. [Google Scholar] [CrossRef]
- Donnelly, C.J.; Zhang, P.W.; Pham, J.T.; Haeusler, A.R.; Mistry, N.A.; Vidensky, S.; Daley, E.L.; Poth, E.M.; Hoover, B.; Fines, D.M.; et al. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 2013, 80, 415–428. [Google Scholar] [CrossRef]
- Stepto, A.; Gallo, J.M.; Shaw, C.E.; Hirth, F. Modelling C9ORF72 hexanucleotide repeat expansion in amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. 2014, 127, 377–389. [Google Scholar] [CrossRef]
- Das, S.; Krainer, A.R. Emerging functions of SRSF1, splicing factor and oncoprotein, in RNA metabolism and cancer. Mol. Cancer Res. 2014, 12, 1195–1204. [Google Scholar] [CrossRef]
- Hautbergue, G.M. RNA Nuclear Export: From Neurological Disorders to Cancer. Adv. Exp. Med. Biol. 2017, 1007, 89–109. [Google Scholar]
- Hautbergue, G.M.; Castelli, L.M.; Ferraiuolo, L.; Sanchez-Martinez, A.; Cooper-Knock, J.; Higginbottom, A.; Lin, Y.H.; Bauer, C.S.; Dodd, J.E.; Myszczynska, M.A.; et al. SRSF1-dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat. Commun. 2017, 8, 16063. [Google Scholar] [CrossRef] [PubMed]
- Celona, B.; Dollen, J.V.; Vatsavayai, S.C.; Kashima, R.; Johnson, J.R.; Tang, A.A.; Hata, A.; Miller, B.L.; Huang, E.J.; Krogan, N.J.; et al. Suppression of C9orf72 RNA repeat-induced neurotoxicity by the ALS-associated RNA-binding protein Zfp106. Elife 2017, 6, e19032. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Gao, K.; Jankovic, J. The role of FUS gene variants in neurodegenerative diseases. Nat. Rev. Neurol. 2014, 10, 337–348. [Google Scholar] [CrossRef]
- Celona, B.; Salomonsson, S.E.; Wu, H.; Dang, B.; Kratochvil, H.T.; Clelland, C.D.; DeGrado, W.F.; Black, B.L. Zfp106 binds to G-quadruplex RNAs and inhibits RAN translation and formation of RNA foci caused by G4C2 repeats. Proc. Natl. Acad. Sci. USA 2024, 121, e2220020121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Donnelly, C.J.; Haeusler, A.R.; Grima, J.C.; Machamer, J.B.; Steinwald, P.; Daley, E.L.; Miller, S.J.; Cunningham, K.M.; Vidensky, S.; et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 2015, 525, 56–61. [Google Scholar] [CrossRef]
- Haeusler, A.R.; Donnelly, C.J.; Periz, G.; Simko, E.A.; Shaw, P.G.; Kim, M.S.; Maragakis, N.J.; Troncoso, J.C.; Pandey, A.; Sattler, R.; et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 2014, 507, 195–200. [Google Scholar] [CrossRef]
- Gendron, T.F.; Bieniek, K.F.; Zhang, Y.J.; Jansen-West, K.; Ash, P.E.; Caulfield, T.; Daughrity, L.; Dunmore, J.H.; Castanedes-Casey, M.; Chew, J.; et al. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta Neuropathol. 2013, 126, 829–844. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Jansen-West, K.; Xu, Y.F.; Gendron, T.F.; Bieniek, K.F.; Lin, W.L.; Sasaguri, H.; Caulfield, T.; Hubbard, J.; Daughrity, L.; et al. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta Neuropathol. 2014, 128, 505–524. [Google Scholar] [CrossRef]
- Odeh, H.M.; Shorter, J. Arginine-rich dipeptide-repeat proteins as phase disruptors in C9-ALS/FTD. Emerg. Top. Life Sci. 2020, 4, 293–305. [Google Scholar]
- Cook, C.N.; Wu, Y.; Odeh, H.M.; Gendron, T.F.; Jansen-West, K.; Del Rosso, G.; Yue, M.; Jiang, P.; Gomes, E.; Tong, J.; et al. C9orf72 poly(GR) aggregation induces TDP-43 proteinopathy. Sci. Transl. Med. 2020, 12, eabb3774. [Google Scholar] [CrossRef]
- LaClair, K.D.; Zhou, Q.; Michaelsen, M.; Wefers, B.; Brill, M.S.; Janjic, A.; Rathkolb, B.; Farny, D.; Cygan, M.; de Angelis, M.H.; et al. Congenic expression of poly-GA but not poly-PR in mice triggers selective neuron loss and interferon responses found in C9orf72 ALS. Acta Neuropathol. 2020, 140, 121–142. [Google Scholar] [CrossRef] [PubMed]
- Moens, T.G.; Niccoli, T.; Wilson, K.M.; Atilano, M.L.; Birsa, N.; Gittings, L.M.; Holbling, B.V.; Dyson, M.C.; Thoeng, A.; Neeves, J.; et al. C9orf72 arginine-rich dipeptide proteins interact with ribosomal proteins in vivo to induce a toxic translational arrest that is rescued by eIF1A. Acta Neuropathol. 2019, 137, 487–500. [Google Scholar] [CrossRef]
- Dong, D.; Zhang, Z.; Li, Y.; Latallo, M.J.; Wang, S.; Nelson, B.; Wu, R.; Krishnan, G.; Gao, F.B.; Wu, B.; et al. Poly-GR repeats associated with ALS/FTD gene C9ORF72 impair translation elongation and induce a ribotoxic stress response in neurons. Sci. Signal. 2024, 17, eadl1030. [Google Scholar] [CrossRef]
- Sun, Y.; Eshov, A.; Zhou, J.; Isiktas, A.U.; Guo, J.U. C9orf72 arginine-rich dipeptide repeats inhibit UPF1-mediated RNA decay via translational repression. Nat. Commun. 2020, 11, 3354. [Google Scholar] [CrossRef] [PubMed]
- Andrade, N.S.; Ramic, M.; Esanov, R.; Liu, W.; Rybin, M.J.; Gaidosh, G.; Abdallah, A.; Del’Olio, S.; Huff, T.C.; Chee, N.T.; et al. Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol. Neurodegener. 2020, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef]
- Suzuki, H.; Matsuoka, M. Proline-arginine poly-dipeptide encoded by the C9orf72 repeat expansion inhibits adenosine deaminase acting on RNA. J. Neurochem. 2021, 158, 753–765. [Google Scholar] [CrossRef]
- Hideyama, T.; Yamashita, T.; Aizawa, H.; Tsuji, S.; Kakita, A.; Takahashi, H.; Kwak, S. Profound downregulation of the RNA editing enzyme ADAR2 in ALS spinal motor neurons. Neurobiol. Dis. 2012, 45, 1121–1128. [Google Scholar] [CrossRef]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar]
- Tg, S.; Chase, K.J.; Liu, F.; Lloyd, T.E.; Rossoll, W.; Zhang, K. c-Jun N-terminal Kinase Promotes Stress Granule Assembly and Neurodegeneration in C9orf72-mediated ALS and FTD. J. Neurosci. 2023, 43, 3186–3197. [Google Scholar]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef]
- Jimenez-Villegas, J.; Kirby, J.; Mata, A.; Cadenas, S.; Turner, M.R.; Malaspina, A.; Shaw, P.J.; Cuadrado, A.; Rojo, A.I. Dipeptide Repeat Pathology in C9orf72-ALS Is Associated with Redox, Mitochondrial and NRF2 Pathway Imbalance. Antioxidants 2022, 11, 1897. [Google Scholar] [CrossRef] [PubMed]
- Cicardi, M.E.; Hallgren, J.H.; Mawrie, D.; Krishnamurthy, K.; Markandaiah, S.S.; Nelson, A.T.; Kankate, V.; Anderson, E.N.; Pasinelli, P.; Pandey, U.B.; et al. C9orf72 poly(PR) mediated neurodegeneration is associated with nucleolar stress. bioRxiv 2023, 26, 107505. [Google Scholar] [CrossRef] [PubMed]
- Nicchitta, C.V. An emerging role for the endoplasmic reticulum in stress granule biogenesis. Semin. Cell Dev. Biol. 2024, 156, 160–166. [Google Scholar] [CrossRef]
- Matus, S.; Valenzuela, V.; Medinas, D.B.; Hetz, C. ER Dysfunction and Protein Folding Stress in ALS. Int. J. Cell Biol. 2013, 2013, 674751. [Google Scholar] [CrossRef] [PubMed]
- Sultana, J.; Ragagnin, A.M.G.; Parakh, S.; Saravanabavan, S.; Soo, K.Y.; Vidal, M.; Jagaraj, C.J.; Ding, K.; Wu, S.; Shadfar, S.; et al. C9orf72-Associated Dipeptide Repeat Expansions Perturb ER-Golgi Vesicular Trafficking, Inducing Golgi Fragmentation and ER Stress, in ALS/FTD. Mol. Neurobiol. 2024, 61, 10318–10338. [Google Scholar] [CrossRef]
- Gupta, R.; Lan, M.; Mojsilovic-Petrovic, J.; Choi, W.H.; Safren, N.; Barmada, S.; Lee, M.J.; Kalb, R. The Proline/Arginine Dipeptide from Hexanucleotide Repeat Expanded C9ORF72 Inhibits the Proteasome. Eneuro 2017, 4, ENEURO. [Google Scholar] [CrossRef]
- Zhang, Y.; Nelson, S.C.K.; Viera Ortiz, A.P.; Lee, E.B.; Fairman, R. C9orf72 proline-arginine dipeptide repeats disrupt the proteasome and perturb proteolytic activities. J. Neuropathol. Exp. Neurol. 2023, 82, 901–910. [Google Scholar] [CrossRef]
- Xu, S.; Ma, Q.; Shen, J.; Li, N.; Sun, S.; Wang, N.; Chen, Y.; Dong, C.; Tam, K.Y.; Prehn, J.H.M.; et al. ALS-linked C9orf72 dipeptide repeats inhibit starvation-induced autophagy through modulating BCL2-BECN1 interaction. Acta Pharm. Sin. B 2024, 14, 2026–2038. [Google Scholar] [CrossRef]
- Chew, J.; Gendron, T.F.; Prudencio, M.; Sasaguri, H.; Zhang, Y.J.; Castanedes-Casey, M.; Lee, C.W.; Jansen-West, K.; Kurti, A.; Murray, M.E.; et al. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science 2015, 348, 1151–1154. [Google Scholar] [CrossRef]
- Shu, X.; Wei, C.; Tu, W.Y.; Zhong, K.; Qi, S.; Wang, A.; Bai, L.; Zhang, S.X.; Luo, B.; Xu, Z.Z.; et al. Negative regulation of TREM2-mediated C9orf72 poly-GA clearance by the NLRP3 inflammasome. Cell Rep. 2023, 42, 112133. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Taso, O.; Wang, R.; Bayram, S.; Graham, A.C.; Garcia-Reitboeck, P.; Mallach, A.; Andrews, W.D.; Piers, T.M.; Botia, J.A.; et al. Trem2 promotes anti-inflammatory responses in microglia and is suppressed under pro-inflammatory conditions. Hum. Mol. Genet. 2020, 29, 3224–3248. [Google Scholar] [CrossRef]
- Rivers-Auty, J.; Hoyle, C.; Pointer, A.; Lawrence, C.; Pickering-Brown, S.; Brough, D.; Ryan, S. C9orf72 dipeptides activate the NLRP3 inflammasome. Brain Commun. 2024, 6, fcae282. [Google Scholar] [CrossRef]
- Jafarinia, H.; van der Giessen, E.; Onck, P.R. C9orf72 polyPR directly binds to various nuclear transport components. Elife 2024, 12, RP89694. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, B.; LaClair, K.D.; Riemenschneider, H.; Zhou, Q.; Frottin, F.; Mareljic, N.; Czuppa, M.; Farny, D.; Hartmann, H.; Michaelsen, M.; et al. Cell-to-cell transmission of C9orf72 poly-(Gly-Ala) triggers key features of ALS/FTD. EMBO J. 2020, 39, e102811. [Google Scholar] [CrossRef]
- Geng, Y.; Cai, Q. Role of C9orf72 hexanucleotide repeat expansions in ALS/FTD pathogenesis. Front. Mol. Neurosci. 2024, 17, 1322720. [Google Scholar] [CrossRef]
- Steffke, C.; Agarwal, S.; Kabashi, E.; Catanese, A. Overexpression of Toxic Poly(Glycine-Alanine) Aggregates in Primary Neuronal Cultures Induces Time-Dependent Autophagic and Synaptic Alterations but Subtle Activity Impairments. Cells 2024, 13, 1300. [Google Scholar] [CrossRef]
- Jensen, B.K.; Schuldi, M.H.; McAvoy, K.; Russell, K.A.; Boehringer, A.; Curran, B.M.; Krishnamurthy, K.; Wen, X.; Westergard, T.; Ma, L.; et al. Synaptic dysfunction induced by glycine-alanine dipeptides in C9orf72-ALS/FTD is rescued by SV2 replenishment. EMBO Mol. Med. 2020, 12, e10722. [Google Scholar] [CrossRef]
- Jo, Y.; Lee, J.; Lee, S.Y.; Kwon, I.; Cho, H. Poly-dipeptides produced from C9orf72 hexanucleotide repeats cause selective motor neuron hyperexcitability in ALS. Proc. Natl. Acad. Sci. USA 2022, 119, e2113813119. [Google Scholar] [CrossRef]
- Su, M.Y.; Fromm, S.A.; Zoncu, R.; Hurley, J.H. Structure of the C9orf72 ARF GAP complex that is haploinsufficient in ALS and FTD. Nature 2020, 585, 251–255. [Google Scholar] [CrossRef]
- Tang, D.; Sheng, J.; Xu, L.; Yan, C.; Qi, S. The C9orf72-SMCR8-WDR41 complex is a GAP for small GTPases. Autophagy 2020, 16, 1542–1543. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; MacNair, L.; McLean, J.; McGoldrick, P.; McKeever, P.; Soleimani, S.; Keith, J.; Zinman, L.; Rogaeva, E.; Robertson, J. C9orf72 isoforms in Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. Brain Res. 2016, 1647, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Amick, J.; Roczniak-Ferguson, A.; Ferguson, S.M. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol. Biol. Cell. 2016, 27, 3040–3051. [Google Scholar] [CrossRef]
- Zheng, W.; Wang, K.; Wu, Y.; Yan, G.; Zhang, C.; Li, Z.; Wang, L.; Chen, S. C9orf72 regulates the unfolded protein response and stress granule formation by interacting with eIF2alpha. Theranostics 2022, 12, 7289–7306. [Google Scholar] [CrossRef] [PubMed]
- Ciura, S.; Lattante, S.; Le Ber, I.; Latouche, M.; Tostivint, H.; Brice, A.; Kabashi, E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Ann. Neurol. 2013, 74, 180–187. [Google Scholar] [CrossRef]
- Therrien, M.; Rouleau, G.A.; Dion, P.A.; Parker, J.A. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PLoS ONE 2013, 8, e83450. [Google Scholar] [CrossRef]
- Koppers, M.; Blokhuis, A.M.; Westeneng, H.J.; Terpstra, M.L.; Zundel, C.A.; Vieira de Sa, R.; Schellevis, R.D.; Waite, A.J.; Blake, D.J.; Veldink, J.H.; et al. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann. Neurol. 2015, 78, 426–438. [Google Scholar] [CrossRef]
- Beckers, J.; Tharkeshwar, A.K.; Van Damme, P. C9orf72 ALS-FTD: Recent evidence for dysregulation of the autophagy-lysosome pathway at multiple levels. Autophagy 2021, 17, 3306–3322. [Google Scholar] [CrossRef]
- Diab, R.; Pilotto, F.; Saxena, S. Autophagy and neurodegeneration: Unraveling the role of C9ORF72 in the regulation of autophagy and its relationship to ALS-FTD pathology. Front. Cell Neurosci. 2023, 17, 1086895. [Google Scholar] [CrossRef]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.B.; Chen, H.J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef]
- Sellier, C.; Campanari, M.L.; Julie Corbier, C.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef]
- Teyssou, E.; Takeda, T.; Lebon, V.; Boillee, S.; Doukoure, B.; Bataillon, G.; Sazdovitch, V.; Cazeneuve, C.; Meininger, V.; LeGuern, E.; et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: Genetics and neuropathology. Acta Neuropathol. 2013, 125, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Riemslagh, F.W.; Lans, H.; Seelaar, H.; Severijnen, L.; Melhem, S.; Vermeulen, W.; Aronica, E.; Pasterkamp, R.J.; van Swieten, J.C.; Willemsen, R. HR23B pathology preferentially co-localizes with p62, pTDP-43 and poly-GA in C9ORF72-linked frontotemporal dementia and amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2019, 7, 39. [Google Scholar] [CrossRef]
- Chitiprolu, M.; Jagow, C.; Tremblay, V.; Bondy-Chorney, E.; Paris, G.; Savard, A.; Palidwor, G.; Barry, F.A.; Zinman, L.; Keith, J.; et al. A complex of C9ORF72 and p62 uses arginine methylation to eliminate stress granules by autophagy. Nat. Commun. 2018, 9, 2794. [Google Scholar] [CrossRef] [PubMed]
- Levine, T.P.; Daniels, R.D.; Gatta, A.T.; Wong, L.H.; Hayes, M.J. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics 2013, 29, 499–503. [Google Scholar] [CrossRef]
- Iyer, S.; Subramanian, V.; Acharya, K.R. C9orf72, a protein associated with amyotrophic lateral sclerosis (ALS) is a guanine nucleotide exchange factor. PeerJ 2018, 6, e5815. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.P.; Smith, E.F.; Grierson, A.J.; De Vos, K.J. C9orf72 plays a central role in Rab GTPase-dependent regulation of autophagy. Small GTPases 2018, 9, 399–408. [Google Scholar] [CrossRef]
- Pang, W.L.; Hu, F.H. Cellular and physiological functions of C9ORF72 and implications for ALS/FTD. J. Neurochem. 2021, 157, 334–350. [Google Scholar] [CrossRef]
- Longatti, A.; Lamb, C.A.; Razi, M.; Yoshimura, S.; Barr, F.A.; Tooze, S.A. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J. Cell Biol. 2012, 197, 659–675. [Google Scholar] [CrossRef]
- Fader, C.M.; Sanchez, D.; Furlan, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef]
- Szatmari, Z.; Kis, V.; Lippai, M.; Hegedus, K.; Farago, T.; Lorincz, P.; Tanaka, T.; Juhasz, G.; Sass, M. Rab11 facilitates cross-talk between autophagy and endosomal pathway through regulation of Hook localization. Mol. Biol. Cell. 2014, 25, 522–531. [Google Scholar] [CrossRef]
- Farg, M.A.; Sundaramoorthy, V.; Sultana, J.M.; Yang, S.; Atkinson, R.A.; Levina, V.; Halloran, M.A.; Gleeson, P.A.; Blair, I.P.; Soo, K.Y.; et al. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Hum. Mol. Genet. 2014, 23, 3579–3595. [Google Scholar] [CrossRef] [PubMed]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. mTOR: On target for novel therapeutic strategies in the nervous system. Trends Mol. Med. 2013, 19, 51–60. [Google Scholar] [CrossRef]
- Chong, Z.Z. mTOR: A Novel Therapeutic Target for Diseases of Multiple Systems. Curr. Drug Targets 2015, 16, 1107–1132. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, E. Rag GTPase in amino acid signaling. Amino Acids 2016, 48, 915–928. [Google Scholar] [CrossRef]
- Dossou, A.S.; Basu, A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers 2019, 11, 1422. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, H.; Tao, Z.; Xia, Q.; Hao, Z.; Prehn, J.H.M.; Zhen, X.; Wang, G.; Ying, Z. C9orf72 associates with inactive Rag GTPases and regulates mTORC1-mediated autophagosomal and lysosomal biogenesis. Aging Cell. 2020, 19, e13126. [Google Scholar] [CrossRef]
- Clarke, P.R. Signaling to nuclear transport. Dev. Cell 2008, 14, 316–318. [Google Scholar] [CrossRef]
- McGoldrick, P.; Lau, A.; You, Z.; Durcan, T.M.; Robertson, J. Loss of C9orf72 perturbs the Ran-GTPase gradient and nucleocytoplasmic transport, generating compositionally diverse Importin beta-1 granules. Cell Rep. 2023, 42, 112134. [Google Scholar] [CrossRef]
- Femiano, C.; Bruno, A.; Gilio, L.; Buttari, F.; Dolcetti, E.; Galifi, G.; Azzolini, F.; Borrelli, A.; Furlan, R.; Finardi, A.; et al. Inflammatory signature in amyotrophic lateral sclerosis predicting disease progression. Sci. Rep. 2024, 14, 19796. [Google Scholar] [CrossRef]
- Kuhle, J.; Lindberg, R.L.; Regeniter, A.; Mehling, M.; Steck, A.J.; Kappos, L.; Czaplinski, A. Increased levels of inflammatory chemokines in amyotrophic lateral sclerosis. Eur. J. Neurol. 2009, 16, 771–774. [Google Scholar] [CrossRef]
- Sullivan, P.M.; Zhou, X.; Robins, A.M.; Paushter, D.H.; Kim, D.; Smolka, M.B.; Hu, F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 2016, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Vahsen, B.F.; Nalluru, S.; Morgan, G.R.; Farrimond, L.; Carroll, E.; Xu, Y.; Cramb, K.M.L.; Amein, B.; Scaber, J.; Katsikoudi, A.; et al. C9orf72-ALS human iPSC microglia are pro-inflammatory and toxic to co-cultured motor neurons via MMP9. Nat. Commun. 2023, 14, 5898. [Google Scholar] [CrossRef] [PubMed]
- Atanasio, A.; Decman, V.; White, D.; Ramos, M.; Ikiz, B.; Lee, H.C.; Siao, C.J.; Brydges, S.; LaRosa, E.; Bai, Y.; et al. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci. Rep. 2016, 6, 23204. [Google Scholar] [CrossRef] [PubMed]
- Fang, T.; Je, G.; Pacut, P.; Keyhanian, K.; Gao, J.; Ghasemi, M. Gene Therapy in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2066. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef]
- Liu, Y.J.; Dodart, J.C.; Tran, H.; Berkovitch, S.; Braun, M.; Byrne, M.; Durbin, A.F.; Hu, X.S.; Iwamoto, N.; Jang, H.G.; et al. Variant-selective stereopure oligonucleotides protect against pathologies associated with -repeat expansion in preclinical models. Nat. Commun. 2021, 12, 847. [Google Scholar] [CrossRef]
- van den Berg, L.H.; Rothstein, J.D.; Shaw, P.J.; Babu, S.; Benatar, M.; Bucelli, R.C.; Genge, A.; Glass, J.D.; Hardiman, O.; Libri, V.; et al. Safety, tolerability, and pharmacokinetics of antisense oligonucleotide BIIB078 in adults with C9orf72-associated amyotrophic lateral sclerosis: A phase 1, randomised, double blinded, placebo-controlled, multiple ascending dose study. Lancet Neurol. 2024, 23, 901–912. [Google Scholar] [CrossRef]
- Liu, Y.; Andreucci, A.; Iwamoto, N.; Yin, Y.; Yang, H.; Liu, F.; Bulychev, A.; Hu, X.S.; Lin, X.; Lamore, S.; et al. Preclinical evaluation of WVE-004, aninvestigational stereopure oligonucleotide forthe treatment of C9orf72-associated ALS or FTD. Mol. Ther. Nucleic Acids 2022, 28, 558–570. [Google Scholar] [CrossRef]
- Tran, H.; Moazami, M.P.; Yang, H.; McKenna-Yasek, D.; Douthwright, C.L.; Pinto, C.; Metterville, J.; Shin, M.; Sanil, N.; Dooley, C.; et al. Suppression of mutant C9orf72 expression by a potent mixed backbone antisense oligonucleotide. Nat. Med. 2022, 28, 117–124. [Google Scholar] [CrossRef]
- Meijboom, K.E.; Abdallah, A.; Fordham, N.P.; Nagase, H.; Rodriguez, T.; Kraus, C.; Gendron, T.F.; Krishnan, G.; Esanov, R.; Andrade, N.S.; et al. CRISPR/Cas9-mediated excision of ALS/FTD-causing hexanucleotide repeat expansion in C9ORF72 rescues major disease mechanisms in vivo and in vitro. Nat. Commun. 2022, 13, 6286. [Google Scholar] [CrossRef] [PubMed]
- Ababneh, N.A.; Scaber, J.; Flynn, R.; Douglas, A.; Barbagallo, P.; Candalija, A.; Turner, M.R.; Sims, D.; Dafinca, R.; Cowley, S.A.; et al. Correction of amyotrophic lateral sclerosis related phenotypes in induced pluripotent stem cell-derived motor neurons carrying a hexanucleotide expansion mutation in C9orf72 by CRISPR/Cas9 genome editing using homology-directed repair. Hum. Mol. Genet. 2020, 29, 2200–2217. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhao, X.F.; Lu, Y.N.; Hayes, L.R.; Wang, J. CRISPR/Cas13d targeting suppresses repeat-associated non-AUG translation of C9orf72 hexanucleotide repeat RNA. J. Clin. Invest. 2024, 134, e179016. [Google Scholar] [CrossRef]
- Cheng, W.; Wang, S.; Zhang, Z.; Morgens, D.W.; Hayes, L.R.; Lee, S.; Portz, B.; Xie, Y.; Nguyen, B.V.; Haney, M.S.; et al. CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron 2019, 104, 885–898. [Google Scholar] [CrossRef]
- Fu, X.; Zhang, Z.; Hayes, L.R.; Wright, N.; Asbury, J.; Li, S.; Ye, Y.; Sun, S. DDX3X overexpression decreases dipeptide repeat proteins in a mouse model of C9ORF72-ALS/FTD. Exp. Neurol. 2024, 376, 114768. [Google Scholar] [CrossRef]
- Licata, N.V.; Cristofani, R.; Salomonsson, S.; Wilson, K.M.; Kempthorne, L.; Vaizoglu, D.; D’Agostino, V.G.; Pollini, D.; Loffredo, R.; Pancher, M.; et al. C9orf72 ALS/FTD dipeptide repeat protein levels are reduced by small molecules that inhibit PKA or enhance protein degradation. EMBO J. 2022, 41, e105026. [Google Scholar] [CrossRef] [PubMed]
- Seidel, M.; Rajkumar, S.; Steffke, C.; Noeth, V.; Agarwal, S.; Roger, K.; Lipecka, J.; Ludolph, A.; Guerrera, C.I.; Boeckers, T.; et al. Propranolol reduces the accumulation of cytotoxic aggregates in C9orf72-ALS/FTD in vitro models. Curr. Res. Neurobiol. 2023, 5, 100105. [Google Scholar] [CrossRef] [PubMed]
- Premasiri, A.S.; Gill, A.L.; Vieira, F.G. Type I PRMT Inhibition Protects Against C9ORF72 Arginine-Rich Dipeptide Repeat Toxicity. Front. Pharmacol. 2020, 11, 569661. [Google Scholar] [CrossRef]
- Dane, T.L.; Gill, A.L.; Vieira, F.G.; Denton, K.R. Reduced C9orf72 expression exacerbates polyGR toxicity in patient iPSC-derived motor neurons and a Type I protein arginine methyltransferase inhibitor reduces that toxicity. Front. Cell Neurosci. 2023, 17, 1134090. [Google Scholar] [CrossRef]
- Xu, W.; Bao, P.; Jiang, X.; Wang, H.; Qin, M.; Wang, R.; Wang, T.; Yang, Y.; Lorenzini, I.; Liao, L.; et al. Reactivation of nonsense-mediated mRNA decay protects against C9orf72 dipeptide-repeat neurotoxicity. Brain 2019, 142, 1349–1364. [Google Scholar] [CrossRef]
- Zaepfel, B.L.; Zhang, Z.; Maulding, K.; Coyne, A.N.; Cheng, W.; Hayes, L.R.; Lloyd, T.E.; Sun, S.; Rothstein, J.D. UPF1 reduces C9orf72 HRE-induced neurotoxicity in the absence of nonsense-mediated decay dysfunction. Cell Rep. 2021, 34, 108925. [Google Scholar] [CrossRef]
- de Calbiac, H.; Renault, S.; Haouy, G.; Jung, V.; Roger, K.; Zhou, Q.; Campanari, M.L.; Chentout, L.; Demy, D.L.; Marian, A.; et al. Poly-GP accumulation due to C9orf72 loss of function induces motor neuron apoptosis through autophagy and mitophagy defects. Autophagy 2024, 20, 2164–2185. [Google Scholar] [CrossRef]
- Li, C.; Qiao, Y.; Jiang, X.; Liu, L.; Zheng, Y.; Qiu, Y.; Cheng, C.; Zhou, F.; Zhou, Y.; Huang, W.; et al. Discovery of a First-in-Class Degrader for the Lipid Kinase PIKfyve. J. Med. Chem. 2023, 66, 12432–12445. [Google Scholar] [CrossRef]
- Babu, S.; Nicholson, K.A.; Rothstein, J.D.; Swenson, A.; Sampognaro, P.J.; Pant, P.; Macklin, E.A.; Spruill, S.; Paganoni, S.; Gendron, T.F.; et al. Apilimod dimesylate in C9orf72 amyotrophic lateral sclerosis: A randomized phase 2a clinical trial. Brain 2024, 147, 2998–3008. [Google Scholar] [CrossRef]
- Tu, W.Y.; Xu, W.; Zhang, J.; Qi, S.; Bai, L.; Shen, C.; Zhang, K. C9orf72 poly-GA proteins impair neuromuscular transmission. Zool. Res. 2023, 44, 331–340. [Google Scholar] [CrossRef]
- Kim, N.; Stiegler, A.L.; Cameron, T.O.; Hallock, P.T.; Gomez, A.M.; Huang, J.H.; Hubbard, S.R.; Dustin, M.L.; Burden, S.J. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell 2008, 135, 334–342. [Google Scholar] [CrossRef]
- Bezakova, G.; Ruegg, M.A. New insights into the roles of agrin. Nat. Rev. Mol. Cell Biol. 2003, 4, 295–308. [Google Scholar] [CrossRef]
- Cicardi, M.E.; Kankate, V.; Sriramoji, S.; Krishnamurthy, K.; Markandaiah, S.S.; Verdone, B.M.; Girdhar, A.; Nelson, A.; Rivas, L.B.; Boehringer, A.; et al. The nuclear import receptor Kapbeta2 modifies neurotoxicity mediated by poly(GR) in C9orf72-linked ALS/FTD. Commun. Biol. 2024, 7, 376. [Google Scholar] [CrossRef]
- Jafarinia, H.; Van der Giessen, E.; Onck, P.R. Molecular basis of C9orf72 poly-PR interference with the beta-karyopherin family of nuclear transport receptors. Sci. Rep. 2022, 12, 21324. [Google Scholar] [CrossRef]
- Pang, W.; Hu, F. C9ORF72 suppresses JAK-STAT mediated inflammation. iScience 2023, 26, 106579. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chong, Z.Z.; Souayah, N. Targeting Gene C9orf72 Pathogenesis for Amyotrophic Lateral Sclerosis. Int. J. Mol. Sci. 2025, 26, 4276. https://doi.org/10.3390/ijms26094276
Chong ZZ, Souayah N. Targeting Gene C9orf72 Pathogenesis for Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences. 2025; 26(9):4276. https://doi.org/10.3390/ijms26094276
Chicago/Turabian StyleChong, Zhao Zhong, and Nizar Souayah. 2025. "Targeting Gene C9orf72 Pathogenesis for Amyotrophic Lateral Sclerosis" International Journal of Molecular Sciences 26, no. 9: 4276. https://doi.org/10.3390/ijms26094276
APA StyleChong, Z. Z., & Souayah, N. (2025). Targeting Gene C9orf72 Pathogenesis for Amyotrophic Lateral Sclerosis. International Journal of Molecular Sciences, 26(9), 4276. https://doi.org/10.3390/ijms26094276