
Evaluation of Illumina and Oxford Nanopore Sequencing for the Study of DNA Methylation in Alzheimer’s Disease and Frontotemporal Dementia

 ,
,  , , , , , , , , , , , ,
, , , , , , , , , , , ,  ,
,  and
and
Abstract
1. Introduction
2. Sequencing Techniques and Technologies
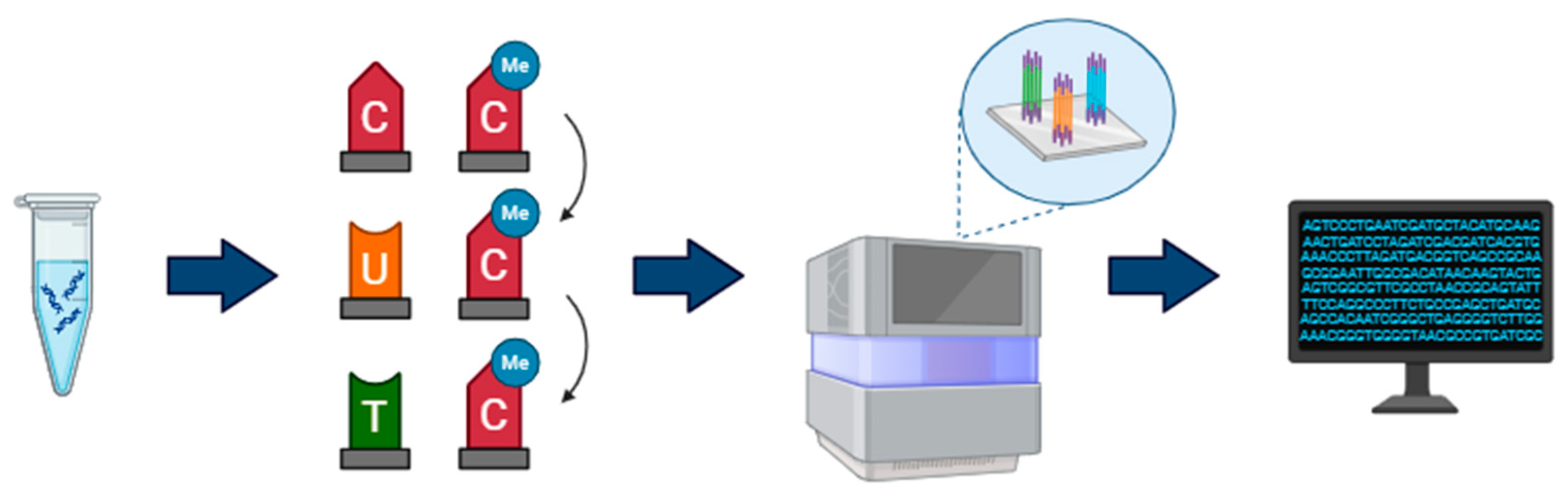
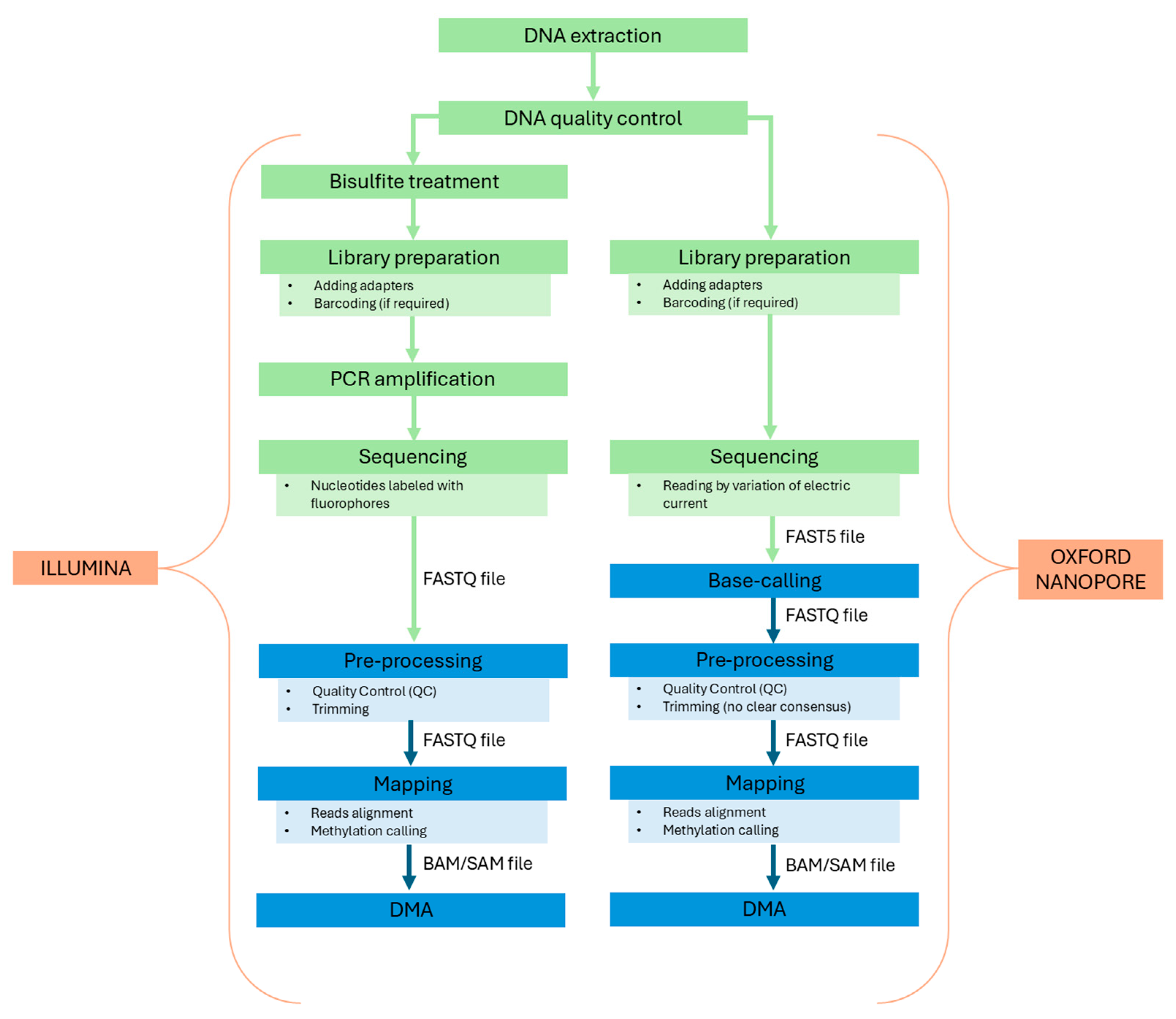
2.1. Illumina Bisulfite Sequencing
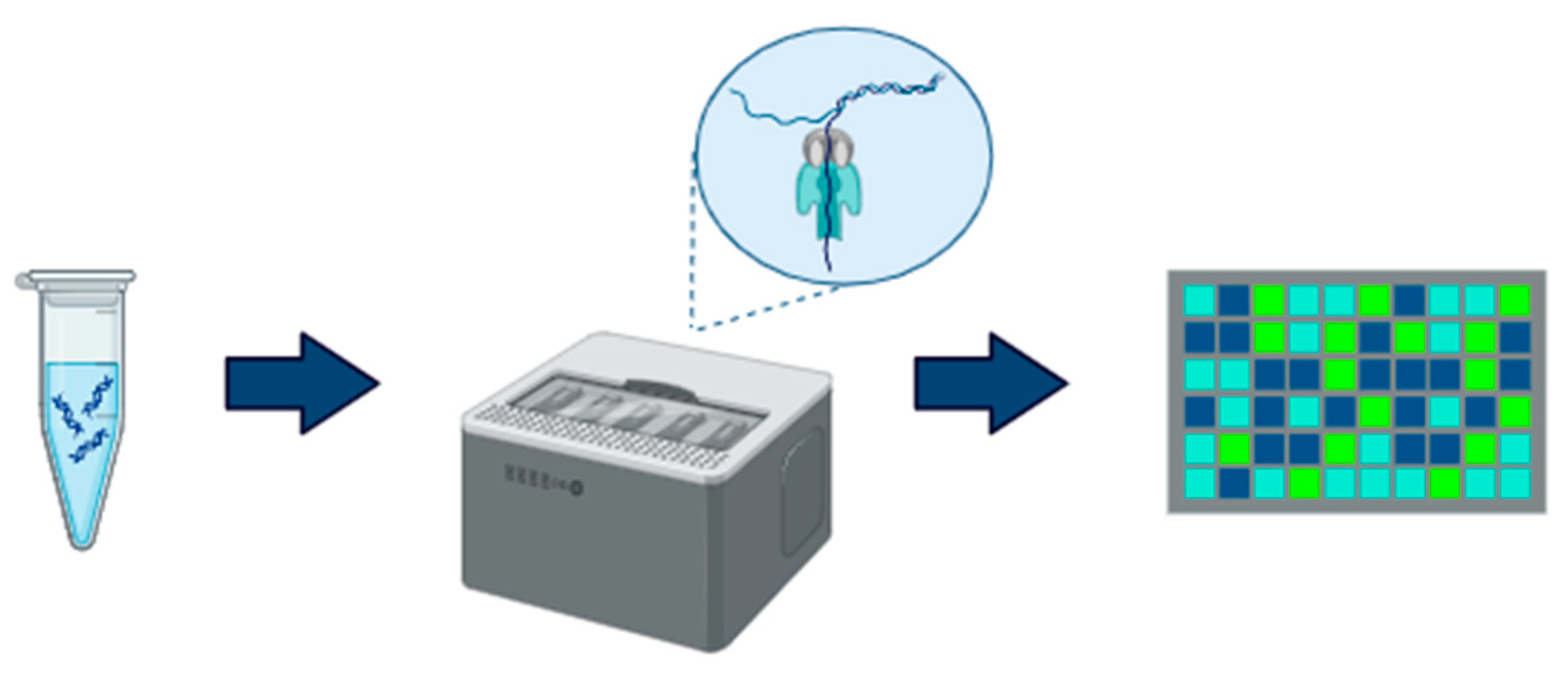
2.2. Oxford Nanopore
3. Comparison
3.1. Accuracy and Efficiency
3.2. Genome Regions
3.3. Costs
3.4. Wet-Lab Protocols
3.5. Bioinformatics Pipelines
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Gibney, E.R.; Nolan, C.M. Epigenetics and Gene Expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Farsetti, A.; Illi, B.; Gaetano, C. How Epigenetics Impacts on Human Diseases. Eur. J. Intern. Med. 2023, 114, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, J.C. Epigenetics in Development. Dev. Dyn. 2007, 236, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA Methylation Patterns and Epigenetic Memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Rasmi, Y.; Shokati, A.; Hassan, A.; Aziz, S.G.; Bastani, S.; Jalali, L.; Moradi, F.; Alipour, S. The Role of DNA Methylation in Progression of Neurological Disorders and Neurodegenerative Diseases as Well as the Prospect of using DNA Methylation Inhibitors as Therapeutic Agents for such Disorders. IBRO Neurosci. Rep. 2022, 14, 28–37. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of Epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The Epigenomics of Cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Krolevets, M.; Cate, V.T.; Prochaska, J.H.; Schulz, A.; Rapp, S.; Tenzer, S.; Andrade-Navarro, M.A.; Horvath, S.; Niehrs, C.; Wild, P.S. DNA Methylation and Cardiovascular Disease in Humans: A Systematic Review and Database of Known CpG Methylation Sites. Clin. Epigenet. 2023, 15, 56. [Google Scholar] [CrossRef]
- Moosavi, A.; Motevalizadeh Ardekani, A. Role of Epigenetics in Biology and Human Diseases. Iran. Biomed. J. 2016, 20, 246–258. [Google Scholar] [CrossRef]
- Vodovotz, Y.; Barnard, N.; Hu, F.B.; Jakicic, J.; Lianov, L.; Loveland, D.; Buysse, D.; Szigethy, E.; Finkel, T.; Sowa, G.; et al. Prioritized Research for the Prevention, Treatment, and Reversal of Chronic Disease: Recommendations from the Lifestyle Medicine Research Summit. Front. Med. 2020, 7, 585744. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.; Gama-Sosa, M.A.; Huang, L.H.; Midgett, R.M.; Kuo, K.C.; McCune, R.A.; Gehrke, C. Amount and Distribution of 5-Methylcytosine in Human DNA from Different Types of Tissues of Cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef]
- Schübeler, D. Function and Information Content of DNA Methylation. Nature 2015, 517, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Illingworth, R.S.; Bird, A.P. CpG Islands—‘A Rough Guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef]
- Long, M.D.; Smiraglia, D.J.; Campbell, M.J. The Genomic Impact of DNA CpG Methylation on Gene Expression; Relationships in Prostate Cancer. Biomolecules 2017, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Boyes, J.; Bird, A. Repression of Genes by DNA Methylation Depends on CpG Density and Promoter Strength: Evidence for Involvement of a Methyl-CpG Binding Protein. EMBO J. 1992, 11, 327–333. [Google Scholar] [CrossRef]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Pääbo, S.; Rebhan, M.; Schübeler, D. Distribution, Silencing Potential and Evolutionary Impact of Promoter DNA Methylation in the Human Genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Jeltsch, A. On the Enzymatic Properties of Dnmt1: Specificity, Processivity, Mechanism of Linear Diffusion and Allosteric Regulation of the Enzyme. Epigenetics 2006, 1, 63–66. [Google Scholar] [CrossRef]
- Ren, W.; Gao, L.; Song, J. Structural Basis of DNMT1 and DNMT3A-Mediated DNA Methylation. Genes 2018, 9, 620. [Google Scholar] [CrossRef]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet Proteins can Convert 5-Methylcytosine to 5-Formylcytosine and 5-Carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-Methylcytosine to 5-Hydroxymethylcytosine in Mammalian DNA by MLL Partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Younesian, S.; Yousefi, A.; Momeny, M.; Ghaffari, S.H.; Bashash, D. The DNA Methylation in Neurological Diseases. Cells 2022, 11, 3439. [Google Scholar] [CrossRef] [PubMed]
- Rascovsky, K.; Salmon, D.P.; Ho, G.J.; Galasko, D.; Peavy, G.M.; Hansen, L.A.; Thal, L.J. Cognitive Profiles Differ in Autopsy-Confirmed Frontotemporal Dementia and AD. Neurology 2002, 58, 1801–1808. [Google Scholar] [CrossRef]
- Stozická, Z.; Zilka, N.; Novák, M. Risk and Protective Factors for Sporadic Alzheimer’s Disease. Acta Virol. 2007, 51, 205–222. [Google Scholar] [PubMed]
- Rosso, S.M.; Landweer, E.; Houterman, M.; Donker Kaat, L.; van Duijn, C.M.; van Swieten, J.C. Medical and Environmental Risk Factors for Sporadic Frontotemporal Dementia: A Retrospective Case-Control Study. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1574–1576. [Google Scholar] [CrossRef]
- Saraceno, C.; Pagano, L.; Laganà, V.; Geviti, A.; Bagnoli, S.; Ingannato, A.; Mazzeo, S.; Longobardi, A.; Fostinelli, S.; Bellini, S.; et al. Mutational Landscape of Alzheimer’s Disease and Frontotemporal Dementia: Regional Variances in Northern, Central, and Southern Italy. Int. J. Mol. Sci. 2024, 25, 7035. [Google Scholar] [CrossRef]
- Fenoglio, C.; Scarpini, E.; Serpente, M.; Galimberti, D. Role of Genetics and Epigenetics in the Pathogenesis of Alzheimer’s Disease and Frontotemporal Dementia. J. Alzheimers Dis. 2018, 62, 913–932. [Google Scholar] [CrossRef]
- Adani, G.; Filippini, T.; Garuti, C.; Malavolti, M.; Vinceti, G.; Zamboni, G.; Tondelli, M.; Galli, C.; Costa, M.; Vinceti, M.; et al. Environmental Risk Factors for Early-Onset Alzheimer’s Dementia and Frontotemporal Dementia: A Case-Control Study in Northern Italy. Int. J. Environ. Res. Public Health 2020, 17, 7941. [Google Scholar] [CrossRef]
- De Plano, L.M.; Saitta, A.; Oddo, S.; Caccamo, A. Epigenetic Changes in Alzheimer’s Disease: DNA Methylation and Histone Modification. Cells 2024, 13, 719. [Google Scholar] [CrossRef]
- Shireby, G.; Dempster, E.L.; Policicchio, S.; Smith, R.G.; Pishva, E.; Chioza, B.; Davies, J.P.; Burrage, J.; Lunnon, K.; Seiler Vellame, D.; et al. DNA Methylation Signatures of Alzheimer’s Disease Neuropathology in the Cortex are Primarily Driven by Variation in Non-Neuronal Cell-Types. Nat. Commun. 2022, 13, 5620–5627. [Google Scholar] [CrossRef]
- Giannini, L.A.A.; Boers, R.G.; van der Ende, E.L.; Poos, J.M.; Jiskoot, L.C.; Boers, J.B.; van IJcken, W.F.J.; Dopper, E.G.; Pijnenburg, Y.A.L.; Seelaar, H.; et al. Distinctive Cell-Free DNA Methylation Characterizes Presymptomatic Genetic Frontotemporal Dementia. Ann. Clin. Transl. Neurol. 2024, 11, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, N.; Evans, A.; Wojta, K.; Yang, Z.; Boks, M.M.; Kahn, R.S.; de Boer, S.C.M.; van der Lee, S.J.; Pijnenburg, Y.A.L.; Reus, L.M.; et al. Accurate DNA Methylation Predictor for C9orf72 Repeat Expansion Alleles in the Pathogenic Range. bioRxiv 2025, Preprint. [Google Scholar] [CrossRef]
- Macías, M.; Alba-Linares, J.J.; Acha, B.; Blanco-Luquin, I.; Fernández, A.F.; Álvarez-Jiménez, J.; Urdánoz-Casado, A.; Roldan, M.; Robles, M.; Cabezon-Arteta, E.; et al. Advancing Personalized Medicine in Alzheimer’s Disease: Liquid Biopsy Epigenomics Unveil APOE Ε4-Linked Methylation Signatures. Int. J. Mol. Sci. 2025, 26, 3419. [Google Scholar] [CrossRef]
- Harvey, J.; Imm, J.; Kouhsar, M.; Smith, A.R.; Creese, B.; Smith, R.G.; Wheildon, G.; Chouliaras, L.; Shireby, G.; Jaunmuktane, Z.; et al. Interrogating DNA Methylation Associated with Lewy Body Pathology in a Cross Brain-Region and Multi-Cohort Study. medRxiv 2025, Preprint. [Google Scholar] [CrossRef]
- Zhang, W.; Young, J.I.; Gomez, L.; Schmidt, M.A.; Lukacsovich, D.; Kunkle, B.W.; Chen, X.S.; Martin, E.R.; Wang, L. Blood DNA Methylation Signature for Incident Dementia: Evidence from Longitudinal Cohorts. Alzheimers Dement. 2025, 21, e14496. [Google Scholar] [CrossRef]
- Ruan, T.; Ling, Y.; Wu, C.; Niu, Y.; Liu, G.; Xu, C.; Lv, Z.; Yuan, Y.; Zhou, X.; Wang, Q.; et al. Abnormal Epigenetic Modification of Lysosome and Lipid Regulating Genes in Alzheimer’s Disease. J. Alzheimers Dis. 2025, 104, 1185–1200. [Google Scholar] [CrossRef]
- Smith, R.G.; Pishva, E.; Shireby, G.; Smith, A.R.; Roubroeks, J.A.Y.; Hannon, E.; Wheildon, G.; Mastroeni, D.; Gasparoni, G.; Riemenschneider, M.; et al. A Meta-Analysis of Epigenome-Wide Association Studies in Alzheimer’s Disease Highlights Novel Differentially Methylated Loci Across Cortex. Nat. Commun. 2021, 12, 3517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Silva, T.C.; Young, J.I.; Gomez, L.; Schmidt, M.A.; Hamilton-Nelson, K.L.; Kunkle, B.W.; Chen, X.; Martin, E.R.; Wang, L. Epigenome-Wide Meta-Analysis of DNA Methylation Differences in Prefrontal Cortex Implicates the Immune Processes in Alzheimer’s Disease. Nat. Commun. 2020, 11, 6114. [Google Scholar] [CrossRef]
- Taskesen, E.; Mishra, A.; van der Sluis, S.; Ferrari, R.; International FTD-Genomics Consortium; Veldink, J.H.; van Es, M.A.; Smit, A.B.; Posthuma, D.; Pijnenburg, Y. Susceptible Genes and Disease Mechanisms Identified in Frontotemporal Dementia and Frontotemporal Dementia with Amyotrophic Lateral Sclerosis by DNA-Methylation and GWAS. Sci. Rep. 2017, 7, 8899. [Google Scholar] [CrossRef]
- Fodder, K.; Murthy, M.; Rizzu, P.; Toomey, C.E.; Hasan, R.; Humphrey, J.; Raj, T.; Lunnon, K.; Mill, J.; Heutink, P.; et al. Brain DNA Methylomic Analysis of Frontotemporal Lobar Degeneration Reveals OTUD4 in Shared Dysregulated Signatures Across Pathological Subtypes. Acta Neuropathol. 2023, 146, 77–95. [Google Scholar] [CrossRef]
- Park, H.; Shin, J.; Kim, Y.; Saito, T.; Saido, T.C.; Kim, J. CRISPR/dCas9-Dnmt3a-Mediated Targeted DNA Methylation of APP Rescues Brain Pathology in a Mouse Model of Alzheimer’s Disease. Transl. Neurodegener 2022, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Nagata, K.; Hatsuta, H.; Takuma, H.; Bundo, M.; Iwamoto, K.; Tamaoka, A.; Murayama, S.; Saido, T.; Tsuji, S. Altered CpG Methylation in Sporadic Alzheimer’s Disease is Associated with APP and MAPT Dysregulation. Hum. Mol. Genet. 2014, 23, 648–656. [Google Scholar] [CrossRef]
- Monti, N.; Cavallaro, R.A.; Stoccoro, A.; Nicolia, V.; Scarpa, S.; Kovacs, G.G.; Fiorenza, M.T.; Lucarelli, M.; Aronica, E.; Ferrer, I.; et al. CpG and Non-CpG Presenilin1 Methylation Pattern in Course of Neurodevelopment and Neurodegeneration is Associated with Gene Expression in Human and Murine Brain. Epigenetics 2020, 15, 781–799. [Google Scholar] [CrossRef] [PubMed]
- Mori, H.; Yoshino, Y.; Ueno, M.; Funahashi, Y.; Kumon, H.; Ozaki, Y.; Yamazaki, K.; Ochi, S.; Iga, J.; Ueno, S. Blood MAPT Expression and Methylation Status in Alzheimer’s Disease. PCN Rep. 2022, 1, e65. [Google Scholar] [CrossRef]
- Caillet-Boudin, M.; Buée, L.; Sergeant, N.; Lefebvre, B. Regulation of Human MAPT Gene Expression. Mol. Neurodegener. 2015, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Galimberti, D.; D’Addario, C.; Dell’osso, B.; Fenoglio, C.; Marcone, A.; Cerami, C.; Cappa, S.F.; Palazzo, M.C.; Arosio, B.; Mari, D.; et al. Progranulin Gene (GRN) Promoter Methylation is Increased in Patients with Sporadic Frontotemporal Lobar Degeneration. Neurol. Sci. 2013, 34, 899–903. [Google Scholar] [CrossRef]
- Xi, Z.; Rainero, I.; Rubino, E.; Pinessi, L.; Bruni, A.C.; Maletta, R.G.; Nacmias, B.; Sorbi, S.; Galimberti, D.; Surace, E.I.; et al. Hypermethylation of the CpG-Island Near the C9orf72 G₄C₂-Repeat Expansion in FTLD Patients. Hum. Mol. Genet. 2014, 23, 5630–5637. [Google Scholar] [CrossRef]
- Gijselinck, I.; Van Mossevelde, S.; van der Zee, J.; Sieben, A.; Engelborghs, S.; De Bleecker, J.; Ivanoiu, A.; Deryck, O.; Edbauer, D.; Zhang, M.; et al. The C9orf72 Repeat Size Correlates with Onset Age of Disease, DNA Methylation and Transcriptional Downregulation of the Promoter. Mol. Psychiatry 2016, 21, 1112–1124. [Google Scholar] [CrossRef]
- De Jager, P.L.; Srivastava, G.; Lunnon, K.; Burgess, J.; Schalkwyk, L.C.; Yu, L.; Eaton, M.L.; Keenan, B.T.; Ernst, J.; McCabe, C.; et al. Alzheimer’s Disease: Early Alterations in Brain DNA Methylation at ANK1, BIN1, RHBDF2 and Other Loci. Nat. Neurosci. 2014, 17, 1156–1163. [Google Scholar] [CrossRef]
- Lunnon, K.; Smith, R.; Hannon, E.; De Jager, P.L.; Srivastava, G.; Volta, M.; Troakes, C.; Al-Sarraj, S.; Burrage, J.; Macdonald, R.; et al. Methylomic Profiling Implicates Cortical Deregulation of ANK1 in Alzheimer’s Disease. Nat. Neurosci. 2014, 17, 1164–1170. [Google Scholar] [CrossRef]
- Yu, L.; Chibnik, L.B.; Srivastava, G.P.; Pochet, N.; Yang, J.; Xu, J.; Kozubek, J.; Obholzer, N.; Leurgans, S.E.; Schneider, J.A.; et al. Association of Brain DNA Methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with Pathological Diagnosis of Alzheimer Disease. JAMA Neurol. 2015, 72, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Tollefsbol, T.O. DNA Methylation Methods: Global DNA Methylation and Methylomic Analyses. Methods 2021, 187, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Derbala, D.; Garnier, A.; Bonnet, E.; Deleuze, J.; Tost, J. Whole-Genome Bisulfite Sequencing Protocol for the Analysis of Genome-Wide DNA Methylation and Hydroxymethylation Patterns at Single-Nucleotide Resolution. Methods Mol. Biol. 2024, 2842, 353–382. [Google Scholar] [CrossRef]
- Hook, P.W.; Timp, W. Beyond Assembly: The Increasing Flexibility of Single-Molecule Sequencing Technology. Nat. Rev. Genet. 2023, 24, 627–641. [Google Scholar] [CrossRef]
- Markets and Markets. Available online: https://www.Marketsandmarkets.Com/ResearchInsight/Next-Generation-Sequencing-Ngs-Technologies-Market.Asp (accessed on 17 April 2025).
- Li, Y.; Tollefsbol, T.O. DNA Methylation Detection: Bisulfite Genomic Sequencing Analysis. Methods Mol. Biol. 2011, 791, 11–21. [Google Scholar] [CrossRef]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A Genomic Sequencing Protocol that Yields a Positive Display of 5-Methylcytosine Residues in Individual DNA Strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.; et al. Human DNA Methylomes at Base Resolution show Widespread Epigenomic Differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.D.; Langmead, B.; Irizarry, R.A. BSmooth: From Whole Genome Bisulfite Sequencing Reads to Differentially Methylated Regions. Genome Biol. 2012, 13, R83. [Google Scholar] [CrossRef]
- Beck, D.; Ben Maamar, M.; Skinner, M.K. Genome-Wide CpG Density and DNA Methylation Analysis Method (MeDIP, RRBS, and WGBS) Comparisons. Epigenetics 2022, 17, 518–530. [Google Scholar] [CrossRef]
- Adusumalli, S.; Mohd Omar, M.F.; Soong, R.; Benoukraf, T. Methodological Aspects of Whole-Genome Bisulfite Sequencing Analysis. Brief. Bioinform. 2015, 16, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Yong, W.; Hsu, F.; Chen, P. Profiling Genome-Wide DNA Methylation. Epigenet. Chromatin 2016, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced Representation Bisulfite Sequencing for Comparative High-Resolution DNA Methylation Analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Morison, I.M. Comparison of Alignment Software for Genome-Wide Bisulphite Sequence Data. Nucleic Acids Res. 2012, 40, e79. [Google Scholar] [CrossRef]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-Read Human Genome Sequencing and its Applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Mantere, T.; Kersten, S.; Hoischen, A. Long-Read Sequencing Emerging in Medical Genetics. Front. Genet. 2019, 10, 426. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, Y.; Bollas, A.; Wang, Y.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef]
- Zhang, Y.; Hatakeyama, S.; Yamaguchi, K.; Furukawa, Y.; Miyano, S.; Yamaguchi, R.; Imoto, S. On the Application of BERT Models for Nanopore Methylation Detection. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021. [Google Scholar]
- Yuen, Z.W.; Srivastava, A.; Daniel, R.; McNevin, D.; Jack, C.; Eyras, E. Systematic Benchmarking of Tools for CpG Methylation Detection from Nanopore Sequencing. Nat. Commun. 2021, 12, 3438. [Google Scholar] [CrossRef]
- Rand, A.C.; Jain, M.; Eizenga, J.M.; Musselman-Brown, A.; Olsen, H.E.; Akeson, M.; Paten, B. Mapping DNA Methylation with High-Throughput Nanopore Sequencing. Nat. Methods 2017, 14, 411–413. [Google Scholar] [CrossRef]
- Loose, M.; Malla, S.; Stout, M. Real-Time Selective Sequencing using Nanopore Technology. Nat. Methods 2016, 13, 751–754. [Google Scholar] [CrossRef]
- Liu, X.; Pang, Y.; Shan, J.; Wang, Y.; Zheng, Y.; Xue, Y.; Zhou, X.; Wang, W.; Sun, Y.; Yan, X.; et al. Beyond the Base Pairs: Comparative Genome-Wide DNA Methylation Profiling Across Sequencing Technologies. Brief. Bioinform. 2024, 25, bbae440. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Okamoto, A. Degradation of DNA by Bisulfite Treatment. Bioorg. Med. Chem. Lett. 2007, 17, 1912–1915. [Google Scholar] [CrossRef] [PubMed]
- Raine, A.; Liljedahl, U.; Nordlund, J. Data Quality of Whole Genome Bisulfite Sequencing on Illumina Platforms. PLoS ONE 2018, 13, e0195972. [Google Scholar] [CrossRef]
- Gong, W.; Pan, X.; Xu, D.; Ji, G.; Wang, Y.; Tian, Y.; Cai, J.; Li, J.; Zhang, Z.; Yuan, X. Benchmarking DNA Methylation Analysis of 14 Alignment Algorithms for Whole Genome Bisulfite Sequencing in Mammals. Comput. Struct. Biotechnol. J. 2022, 20, 4704–4716. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Billingsley, K.J.; Mastoras, M.; Meredith, M.; Monlong, J.; Lorig-Roach, R.; Asri, M.; Alvarez Jerez, P.; Malik, L.; Dewan, R.; et al. Scalable Nanopore Sequencing of Human Genomes Provides a Comprehensive View of Haplotype-Resolved Variation and Methylation. Nat. Methods 2023, 20, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Nanoporetech. Available online: https://nanoporetech.com/platform/accuracy (accessed on 17 April 2025).
- Genner, R.; Akeson, S.; Meredith, M.; Jerez, P.A.; Malik, L.; Baker, B.; Miano-Burkhardt, A.; CARD-Long-Read Team; Paten, B.; Billingsley, K.J.; et al. Assessing Methylation Detection for Primary Human Tissue Using Nanopore Sequencing. bioRxiv, 2024; Preprint. [Google Scholar] [CrossRef]
- Huang, Y.; Pastor, W.A.; Shen, Y.; Tahiliani, M.; Liu, D.R.; Rao, A. The Behaviour of 5-Hydroxymethylcytosine in Bisulfite Sequencing. PLoS ONE 2010, 5, e8888. [Google Scholar] [CrossRef]
- Ebbert, M.T.W.; Jensen, T.D.; Jansen-West, K.; Sens, J.P.; Reddy, J.S.; Ridge, P.G.; Kauwe, J.S.K.; Belzil, V.; Pregent, L.; Carrasquillo, M.M.; et al. Systematic Analysis of Dark and Camouflaged Genes Reveals Disease-Relevant Genes Hiding in Plain Sight. Genome Biol. 2019, 20, 97. [Google Scholar] [CrossRef]
- Liao, X.; Zhu, W.; Zhou, J.; Li, H.; Xu, X.; Zhang, B.; Gao, X. Repetitive DNA Sequence Detection and its Role in the Human Genome. Commun. Biol. 2023, 6, 954. [Google Scholar] [CrossRef]
- Suzuki, M.; Liao, W.; Wos, F.; Johnston, A.D.; DeGrazia, J.; Ishii, J.; Bloom, T.; Zody, M.C.; Germer, S.; Greally, J.M. Whole-Genome Bisulfite Sequencing with Improved Accuracy and Cost. Genome Res. 2018, 28, 1364–1371. [Google Scholar] [CrossRef]
- Gu, H.; Smith, Z.D.; Bock, C.; Boyle, P.; Gnirke, A.; Meissner, A. Preparation of Reduced Representation Bisulfite Sequencing Libraries for Genome-Scale DNA Methylation Profiling. Nat. Protoc. 2011, 6, 468–481. [Google Scholar] [CrossRef]
- Nanoporetech. Available online: https://nanoporetech.com/document/input-dna-rna-qc (accessed on 17 April 2025).
- Zymoresearch. Available online: https://zymoresearch.eu/pages/bisulfite-beginner-guide?srsltid=AfmBOop4jvOAIPlX9lDmWCdHMu3q-7J9ZxxXIJf26R7IV81H3vZiwpfr (accessed on 17 April 2025).
- Gong, T.; Borgard, H.; Zhang, Z.; Chen, S.; Gao, Z.; Deng, Y. Analysis and Performance Assessment of the Whole Genome Bisulfite Sequencing Data Workflow: Currently Available Tools and a Practical Guide to Advance DNA Methylation Studies. Small Methods 2022, 6, e2101251. [Google Scholar] [CrossRef] [PubMed]
- Cd-Genomics. Available online: https://www.cd-genomics.com/overview-of-rrbs-data-analysis-pipeline-alignment-tools-databases-and-challenges.html (accessed on 17 April 2025).
- Shafi, A.; Mitrea, C.; Nguyen, T.; Draghici, S. A Survey of the Approaches for Identifying Differential Methylation using Bisulfite Sequencing Data. Brief. Bioinform. 2018, 19, 737–753. [Google Scholar] [CrossRef]
- Martin, M. CUTADAPT Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef]
- Chen, P.; Cokus, S.J.; Pellegrini, M. BS Seeker: Precise Mapping for Bisulfite Sequencing. BMC Bioinform. 2010, 11, 203. [Google Scholar] [CrossRef] [PubMed]
- Harris, E.Y.; Ponts, N.; Levchuk, A.; Roch, K.L.; Lonardi, S. BRAT: Bisulfite-Treated Reads Analysis Tool. Bioinformatics 2010, 26, 572–573. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole Genome Bisulfite Sequence MAPping Program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef]
- Xi, Y.; Bock, C.; Müller, F.; Sun, D.; Meissner, A.; Li, W. RRBSMAP: A Fast, Accurate and User-Friendly Alignment Tool for Reduced Representation Bisulfite Sequencing. Bioinformatics 2012, 28, 430–432. [Google Scholar] [CrossRef]
- Wu, T.D.; Nacu, S. Fast and SNP-Tolerant Detection of Complex Variants and Splicing in Short Reads. Bioinformatics 2010, 26, 873–881. [Google Scholar] [CrossRef]
- Harris, E.Y.; Ponts, N.; Le Roch, K.G.; Lonardi, S. BRAT-BW: Efficient and Accurate Mapping of Bisulfite-Treated Reads. Bioinformatics 2012, 28, 1795–1796. [Google Scholar] [CrossRef]
- Bock, C.; Reither, S.; Mikeska, T.; Paulsen, M.; Walter, J.; Lengauer, T. BiQ Analyzer: Visualization and Quality Control for DNA Methylation Data from Bisulfite Sequencing. Bioinformatics 2005, 21, 4067–4068. [Google Scholar] [CrossRef]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. methylKit: A Comprehensive R Package for the Analysis of Genome-Wide DNA Methylation Profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Figueroa, M.E.; Rozek, L.S.; Sartor, M.A. MethylSig: A Whole Genome DNA Methylation Analysis Pipeline. Bioinformatics 2014, 30, 2414–2422. [Google Scholar] [CrossRef]
- Saito, Y.; Mituyama, T. Detection of Differentially Methylated Regions from Bisulfite-Seq Data by Hidden Markov Models Incorporating Genome-Wide Methylation Level Distributions. BMC Genom. 2015, 16 (Suppl. S12), S3. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, H.; Lv, J.; Xiao, X.; Zhu, J.; Liu, X.; Su, J.; Li, X.; Wu, Q.; Wang, F.; et al. QDMR: A Quantitative Method for Identification of Differentially Methylated Regions by Entropy. Nucleic Acids Res. 2011, 39, e58. [Google Scholar] [CrossRef] [PubMed]
- Jühling, F.; Kretzmer, H.; Bernhart, S.H.; Otto, C.; Stadler, P.F.; Hoffmann, S. Metilene: Fast and Sensitive Calling of Differentially Methylated Regions from Bisulfite Sequencing Data. Genome Res. 2016, 26, 256–262. [Google Scholar] [CrossRef]
- Yu, X.; Sun, S. HMM-DM: Identifying Differentially Methylated Regions using a Hidden Markov Model. Stat. Appl. Genet. Mol. Biol. 2016, 15, 69–81. [Google Scholar] [CrossRef]
- Warden, C.D.; Lee, H.; Tompkins, J.D.; Li, X.; Wang, C.; Riggs, A.D.; Yu, H.; Jove, R.; Yuan, Y. COHCAP: An Integrative Genomic Pipeline for Single-Nucleotide Resolution DNA Methylation Analysis. Nucleic Acids Res. 2019, 47, 8335–8336. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of Neural Network Basecalling Tools for Oxford Nanopore Sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise Alignment for Nucleotide Sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Liu, Y.; Rosikiewicz, W.; Pan, Z.; Jillette, N.; Wang, P.; Taghbalout, A.; Foox, J.; Mason, C.; Carroll, M.; Cheng, A.; et al. DNA Methylation-Calling Tools for Oxford Nanopore Sequencing: A Survey and Human Epigenome-Wide Evaluation. Genome Biol. 2021, 22, 295. [Google Scholar] [CrossRef]
- Snajder, R.; Leger, A.; Stegle, O.; Bonder, M.J. pycoMeth: A Toolbox for Differential Methylation Testing from Nanopore Methylation Calls. Genome Biol. 2023, 24, 83. [Google Scholar] [CrossRef] [PubMed]
- Bush, S.J. Read Trimming has Minimal Effect on Bacterial SNP-Calling Accuracy. Microb. Genom. 2020, 6, mgen000434. [Google Scholar] [CrossRef]
- Bonenfant, Q.; Noé, L.; Touzet, H. Porechop_ABI: Discovering Unknown Adapters in Oxford Nanopore Technology Sequencing Reads for Downstream Trimming. Bioinform. Adv. 2022, 3, vbac085. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Nguyen, L.T.; Hayes, B.J.; Ross, E.M. Prowler: A Novel Trimming Algorithm for Oxford Nanopore Sequence Data. Bioinformatics 2021, 37, 3936–3937. [Google Scholar] [CrossRef]
- Simpson, J.T.; Workman, R.E.; Zuzarte, P.C.; David, M.; Dursi, L.J.; Timp, W. Detecting DNA Cytosine Methylation using Nanopore Sequencing. Nat. Methods 2017, 14, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Zhang, Y.; Mead, E.A.; Chen, H.; Loo, C.E.; Fan, Y.; Ni, M.; Zhang, X.; Kohli, R.M.; Fang, G. Critical Assessment of Nanopore Sequencing for the Detection of Multiple Forms of DNA Modifications. bioRxiv, 2024; Preprint. [Google Scholar] [CrossRef]
- Bai, X.; Yao, H.; Wu, B.; Liu, L.; Ding, Y.; Xiao, C. DeepBAM: A High-Accuracy Single-Molecule CpG Methylation Detection Tool for Oxford Nanopore Sequencing. Brief. Bioinform. 2024, 25, bbae413. [Google Scholar] [CrossRef]
- Gouil, Q.; Keniry, A. Latest Techniques to Study DNA Methylation. Essays Biochem. 2019, 63, 639–648. [Google Scholar] [CrossRef]
- Xu, L.; Seki, M. Recent Advances in the Detection of Base Modifications using the Nanopore Sequencer. J. Hum. Genet. 2020, 65, 25–33. [Google Scholar] [CrossRef]
- Affinito, O.; Palumbo, D.; Fierro, A.; Cuomo, M.; De Riso, G.; Monticelli, A.; Miele, G.; Chiariotti, L.; Cocozza, S. Nucleotide Distance Influences Co-Methylation between Nearby CpG Sites. Genomics 2020, 112, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Taryma-Leśniak, O.; Bińkowski, J.; Przybylowicz, P.K.; Sokolowska, K.E.; Borowski, K.; Wojdacz, T.K. Methylation Patterns at the Adjacent CpG Sites within Enhancers are a Part of Cell Identity. Epigenet. Chromatin 2024, 17, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic Dysregulation of Enhancers in Neurons is Associated with Alzheimer’s Disease Pathology and Cognitive Symptoms. Nat. Commun. 2019, 10, 2246–2247. [Google Scholar] [CrossRef] [PubMed]
- Kozlenkov, A.; Roussos, P.; Timashpolsky, A.; Barbu, M.; Rudchenko, S.; Bibikova, M.; Klotzle, B.; Byne, W.; Lyddon, R.; Di Narzo, A.F.; et al. Differences in DNA Methylation between Human Neuronal and Glial Cells are Concentrated in Enhancers and Non-CpG Sites. Nucleic Acids Res. 2014, 42, 109–127. [Google Scholar] [CrossRef]
- Rizzardi, L.F.; Hickey, P.F.; Rodriguez DiBlasi, V.; Tryggvadóttir, R.; Callahan, C.M.; Idrizi, A.; Hansen, K.D.; Feinberg, A.P. Neuronal Brain-Region-Specific DNA Methylation and Chromatin Accessibility are Associated with Neuropsychiatric Trait Heritability. Nat. Neurosci. 2019, 22, 307–316. [Google Scholar] [CrossRef]
- Rizzardi, L.F.; Hickey, P.F.; Idrizi, A.; Tryggvadóttir, R.; Callahan, C.M.; Stephens, K.E.; Taverna, S.D.; Zhang, H.; Ramazanoglu, S.; GTEx Consortium; et al. Human Brain Region-Specific Variably Methylated Regions are Enriched for Heritability of Distinct Neuropsychiatric Traits. Genome Biol. 2021, 22, 116. [Google Scholar] [CrossRef]
- Khoodoruth, M.A.S.; Chut-Kai Khoodoruth, W.N.; Al Alwani, R. Exploring the Epigenetic Landscape: The Role of 5-Hydroxymethylcytosine in Neurodevelopmental Disorders. Camb. Prism. Precis. Med. 2024, 2, e5. [Google Scholar] [CrossRef]
- Zhao, J.; Gu, T.; Gao, C.; Miao, G.; Palma-Gudiel, H.; Yu, L.; Yang, J.; Wang, Y.; Li, Y.; Lim, J.; et al. Brain 5-Hydroxymethylcytosine Alterations are Associated with Alzheimer’s Disease Neuropathology. Nat. Commun. 2025, 16, 2842. [Google Scholar] [CrossRef]
- Kuehner, J.N.; Chen, J.; Bruggeman, E.C.; Wang, F.; Li, Y.; Xu, C.; McEachin, Z.T.; Li, Z.; Chen, L.; Hales, C.M.; et al. 5-Hydroxymethylcytosine is Dynamically Regulated during Forebrain Organoid Development and Aberrantly Altered in Alzheimer’s Disease. Cell. Rep. 2021, 35, 109042. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Li, L.; Xu, K.; Ma, Z.; Chow, H.; Herrup, K.; Li, J. Selective Loss of 5hmC Promotes Neurodegeneration in the Mouse Model of Alzheimer’s Disease. FASEB J. 2020, 34, 16364–16382. [Google Scholar] [CrossRef]
- Song, C.; Szulwach, K.E.; Fu, Y.; Dai, Q.; Yi, C.; Li, X.; Li, Y.; Chen, C.; Zhang, W.; Jian, X.; et al. Selective Chemical Labeling Reveals the Genome-Wide Distribution of 5-Hydroxymethylcytosine. Nat. Biotechnol. 2011, 29, 68–72. [Google Scholar] [CrossRef]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global Epigenomic Reconfiguration during Mammalian Brain Development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Mastroeni, D.; McKee, A.; Grover, A.; Rogers, J.; Coleman, P.D. Epigenetic Differences in Cortical Neurons from a Pair of Monozygotic Twins Discordant for Alzheimer’s Disease. PLoS ONE 2009, 4, e6617. [Google Scholar] [CrossRef]
- Condliffe, D.; Wong, A.; Troakes, C.; Proitsi, P.; Patel, Y.; Chouliaras, L.; Fernandes, C.; Cooper, J.; Lovestone, S.; Schalkwyk, L.; et al. Cross-Region Reduction in 5-Hydroxymethylcytosine in Alzheimer’s Disease Brain. Neurobiol. Aging 2014, 35, 1850–1854. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, N.; Dieriks, B.V.; Lill, C.; Faull, R.L.M.; Curtis, M.A.; Dragunow, M. Global Changes in DNA Methylation and Hydroxymethylation in Alzheimer’s Disease Human Brain. Neurobiol. Aging 2014, 35, 1334–1344. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Morales, R.; Esteller, M. Opening Up the DNA Methylome of Dementia. Mol. Psychiatry 2017, 22, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, A.S.; Rutledge, J.C.; Medici, V. DNA Methylation Alterations in Alzheimer’s Disease. Environ. Epigenet. 2017, 3, dvx008. [Google Scholar] [CrossRef]
- Smith, A.R.; Smith, R.G.; Pishva, E.; Hannon, E.; Roubroeks, J.A.Y.; Burrage, J.; Troakes, C.; Al-Sarraj, S.; Sloan, C.; Mill, J.; et al. Parallel Profiling of DNA Methylation and Hydroxymethylation Highlights Neuropathology-Associated Epigenetic Variation in Alzheimer’s Disease. Clin. Epigenet. 2019, 11, 52. [Google Scholar] [CrossRef]
- Ellison, E.M.; Abner, E.L.; Lovell, M.A. Multiregional Analysis of Global 5-Methylcytosine and 5-Hydroxymethylcytosine Throughout the Progression of Alzheimer’s Disease. J. Neurochem. 2017, 140, 383–394. [Google Scholar] [CrossRef]
- Giesselmann, P.; Brändl, B.; Raimondeau, E.; Bowen, R.; Rohrandt, C.; Tandon, R.; Kretzmer, H.; Assum, G.; Galonska, C.; Siebert, R.; et al. Analysis of Short Tandem Repeat Expansions and their Methylation State with Nanopore Sequencing. Nat. Biotechnol. 2019, 37, 1478–1481. [Google Scholar] [CrossRef]
- Ramirez, P.; Sun, W.; Kazempour Dehkordi, S.; Zare, H.; Fongang, B.; Bieniek, K.F.; Frost, B. Nanopore-Based DNA Long-Read Sequencing Analysis of the Aged Human Brain. bioRxiv 2024, Preprint. [Google Scholar] [CrossRef]
- Black, E.M.; Giunta, S. Repetitive Fragile Sites: Centromere Satellite DNA as a Source of Genome Instability in Human Diseases. Genes 2018, 9, 615. [Google Scholar] [CrossRef] [PubMed]
- Barra, V.; Fachinetti, D. The Dark Side of Centromeres: Types, Causes and Consequences of Structural Abnormalities Implicating Centromeric DNA. Nat. Commun. 2018, 9, 4340. [Google Scholar] [CrossRef] [PubMed]
- Udine, E.; DeJesus-Hernandez, M.; Tian, S.; das Neves, S.P.; Crook, R.; Finch, N.A.; Baker, M.C.; Pottier, C.; Graff-Radford, N.R.; Boeve, B.F.; et al. Abundant Transcriptomic Alterations in the Human Cerebellum of Patients with a C9orf72 Repeat Expansion. Acta Neuropathol. 2024, 147, 73. [Google Scholar] [CrossRef]
- Dickson, D.W.; Baker, M.C.; Jackson, J.L.; DeJesus-Hernandez, M.; Finch, N.A.; Tian, S.; Heckman, M.G.; Pottier, C.; Gendron, T.F.; Murray, M.E.; et al. Extensive Transcriptomic Study Emphasizes Importance of Vesicular Transport in C9orf72 Expansion Carriers. Acta Neuropathol. Commun. 2019, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Humphrey, J.; Bettencourt, C.; Newcombe, J.; NYGC ALS Consortium; Lashley, T.; Fratta, P.; Raj, T. Transcriptomic Analysis of Frontotemporal Lobar Degeneration with TDP-43 Pathology Reveals Cellular Alterations Across Multiple Brain Regions. Acta Neuropathol. 2022, 143, 383–401. [Google Scholar] [CrossRef]
- Udine, E.; Finch, N.A.; DeJesus-Hernandez, M.; Jackson, J.L.; Baker, M.C.; Saravanaperumal, S.A.; Wieben, E.; Ebbert, M.T.W.; Shah, J.; Petrucelli, L.; et al. Targeted Long-Read Sequencing to Quantify Methylation of the C9orf72 Repeat Expansion. Mol. Neurodegener. 2024, 19, 99. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Aspect | Illumina | Oxford Nanopore |
|---|---|---|
| Accuracy | Reaching Q20 and Q30; 5 hmC not detected | Almost Q20; real-time sequencing of 5 mC and 5 hmC |
| Efficiency | Alignment rate exceeding 90%; 26.5 million CpG sites | Alignment rate reaching 96%; 28.8 million CpG sites |
| Genome Regions | Repetitive regions are hard to sequence | Effective in resolving repetitive and dark genomic regions |
| Costs | WGBS~300$ per sample RRBS~200$ per sample | WGM ~ 1000$ per sample |
| Wet-lab protocols | need of bisulfite treatment and PCR; ~100 ng input; DNA fragments 200–400 bp | no need of bisulfite treatment and PCR; ~1 µg input; DNA fragments > 30 kb; 260/280 ratio of 1.8 and 260/230 ratio of 2.0–2.2 |
| Bioinformatics pipelines | Specific tools with extensive testing and benchmarking | Few specific tools developed, especially for DMA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagano, L.; Lagrotteria, D.; Facconi, A.; Saraceno, C.; Longobardi, A.; Bellini, S.; Ingannato, A.; Bagnoli, S.; Ducci, T.; Mingrino, A.; et al. Evaluation of Illumina and Oxford Nanopore Sequencing for the Study of DNA Methylation in Alzheimer’s Disease and Frontotemporal Dementia. Int. J. Mol. Sci. 2025, 26, 4198. https://doi.org/10.3390/ijms26094198
Pagano L, Lagrotteria D, Facconi A, Saraceno C, Longobardi A, Bellini S, Ingannato A, Bagnoli S, Ducci T, Mingrino A, et al. Evaluation of Illumina and Oxford Nanopore Sequencing for the Study of DNA Methylation in Alzheimer’s Disease and Frontotemporal Dementia. International Journal of Molecular Sciences. 2025; 26(9):4198. https://doi.org/10.3390/ijms26094198
Chicago/Turabian StylePagano, Lorenzo, Davide Lagrotteria, Alessandro Facconi, Claudia Saraceno, Antonio Longobardi, Sonia Bellini, Assunta Ingannato, Silvia Bagnoli, Tommaso Ducci, Alessandra Mingrino, and et al. 2025. "Evaluation of Illumina and Oxford Nanopore Sequencing for the Study of DNA Methylation in Alzheimer’s Disease and Frontotemporal Dementia" International Journal of Molecular Sciences 26, no. 9: 4198. https://doi.org/10.3390/ijms26094198
APA StylePagano, L., Lagrotteria, D., Facconi, A., Saraceno, C., Longobardi, A., Bellini, S., Ingannato, A., Bagnoli, S., Ducci, T., Mingrino, A., Laganà, V., Paparazzo, E., Borroni, B., Maletta, R., Nacmias, B., Montesanto, A., & Ghidoni, R. (2025). Evaluation of Illumina and Oxford Nanopore Sequencing for the Study of DNA Methylation in Alzheimer’s Disease and Frontotemporal Dementia. International Journal of Molecular Sciences, 26(9), 4198. https://doi.org/10.3390/ijms26094198