
Mapping the Role of P-gp in Multidrug Resistance: Insights from Recent Structural Studies

Abstract
1. Introduction
2. Structure of P-gp
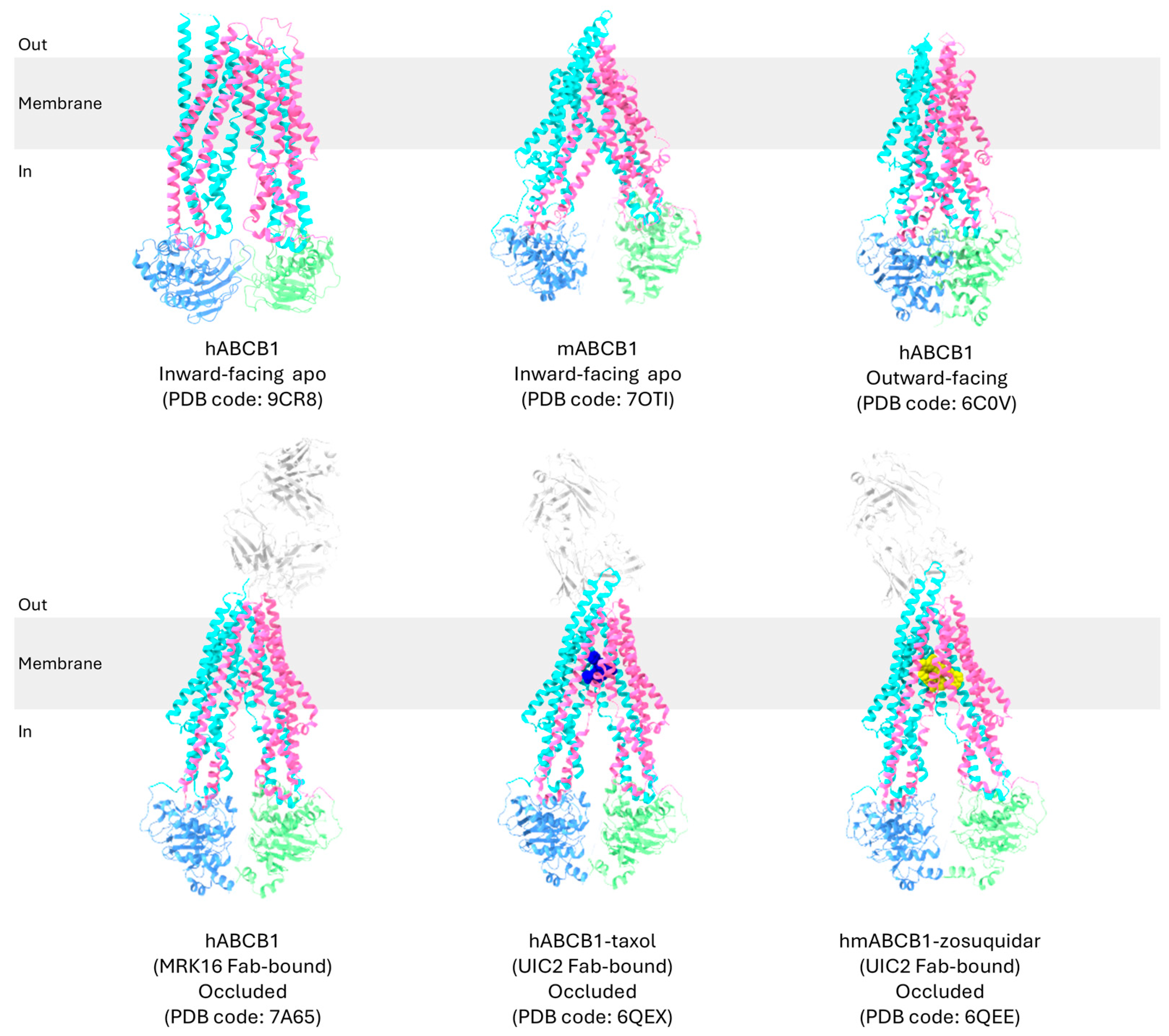
2.1. Overview of High-Resolution Structural Studies of P-gp
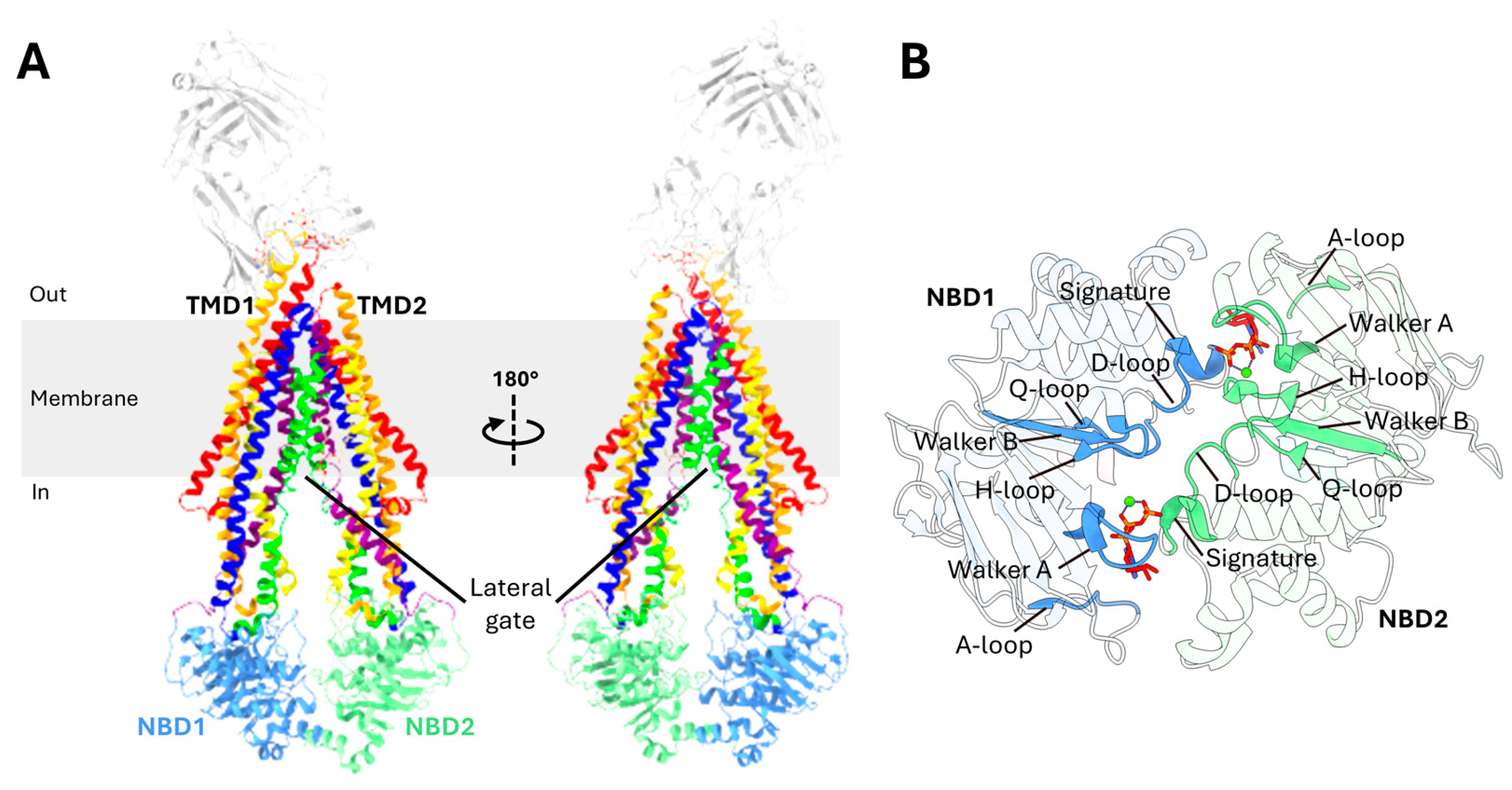
2.2. Domain Organization of P-gp
2.3. Transmembrane Domains (TMDs)
2.4. Nucleotide-Binding Domains (NBDs)
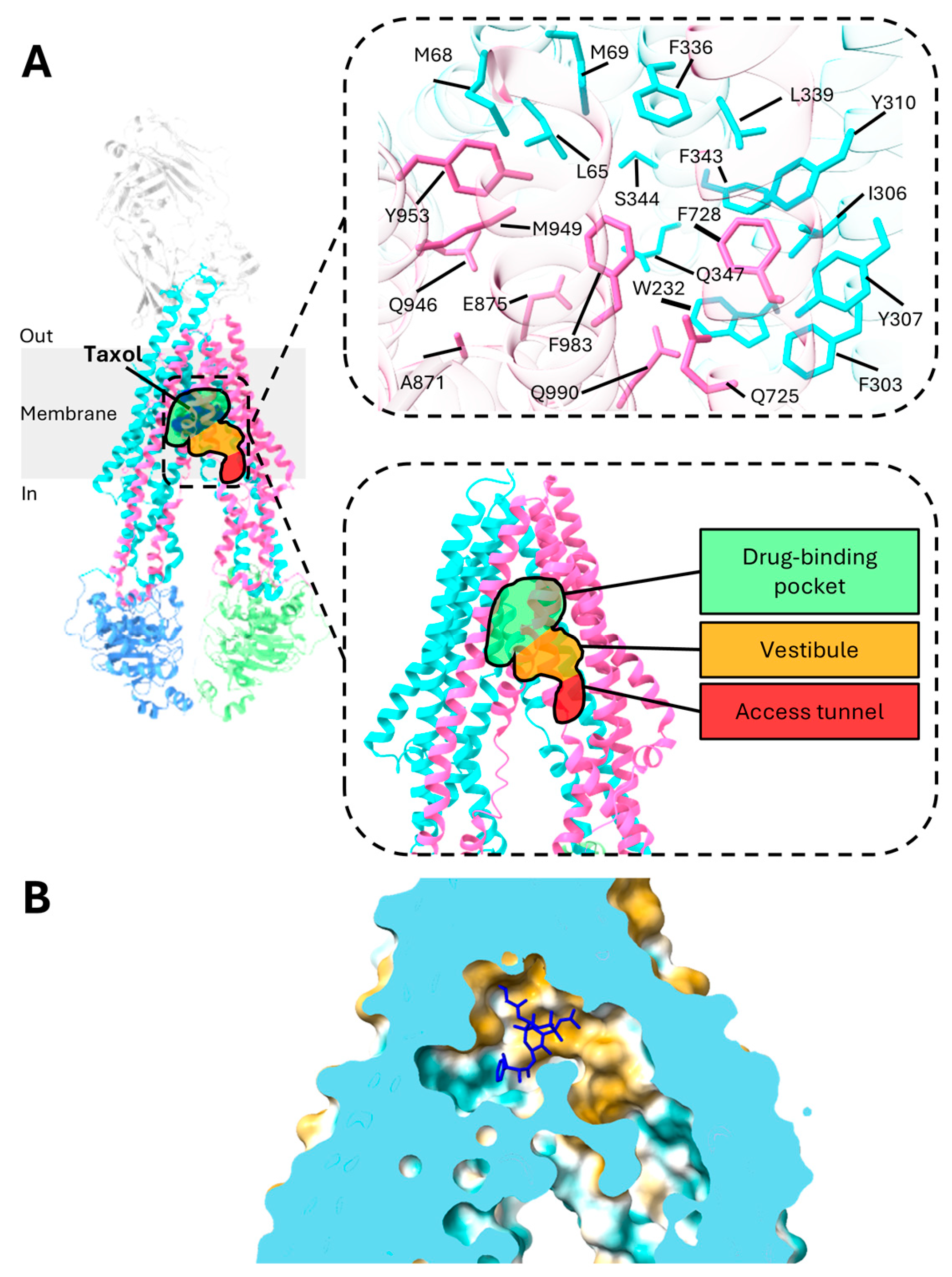
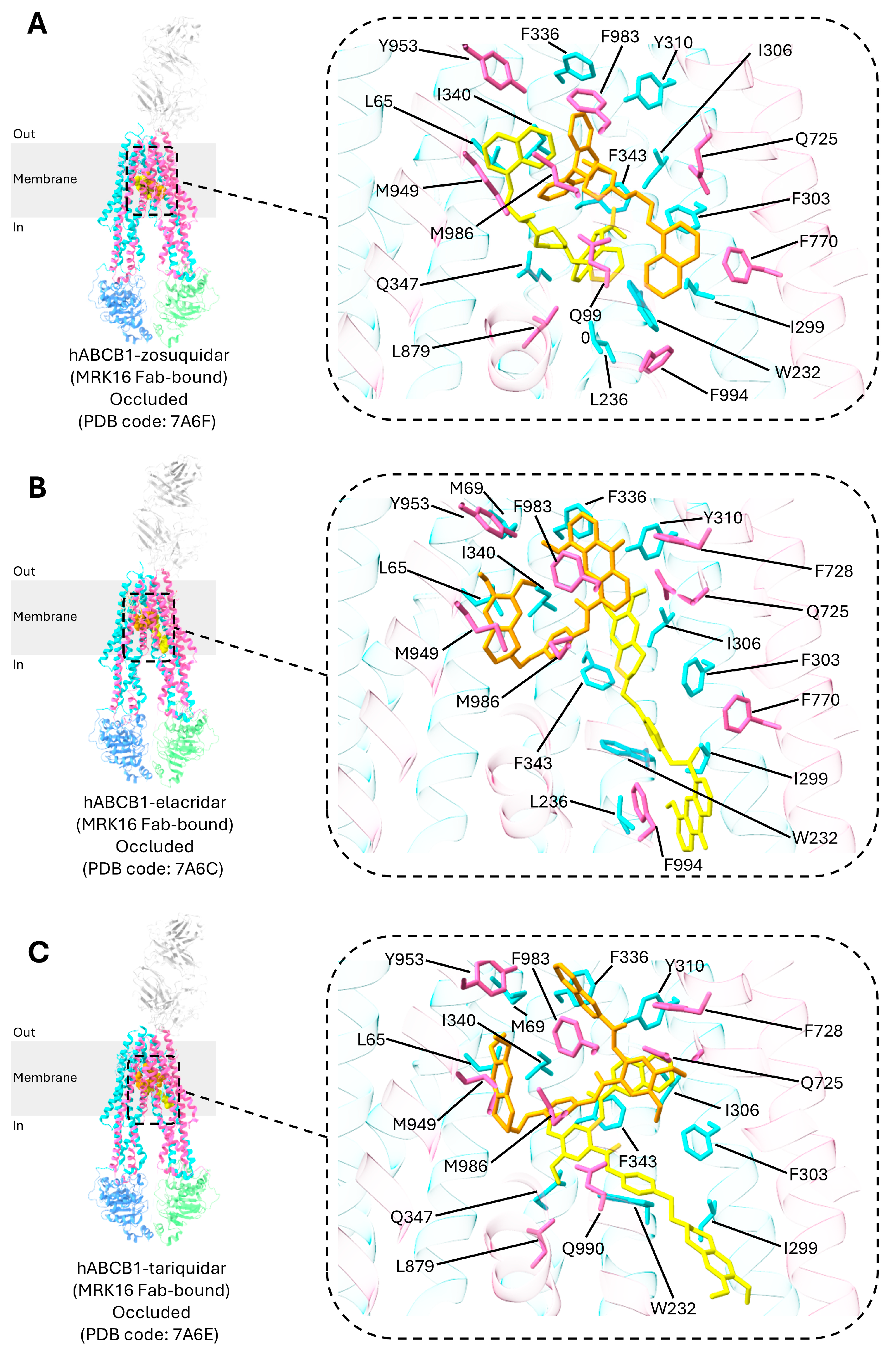
3. Central Drug-Binding Cavity of P-gp
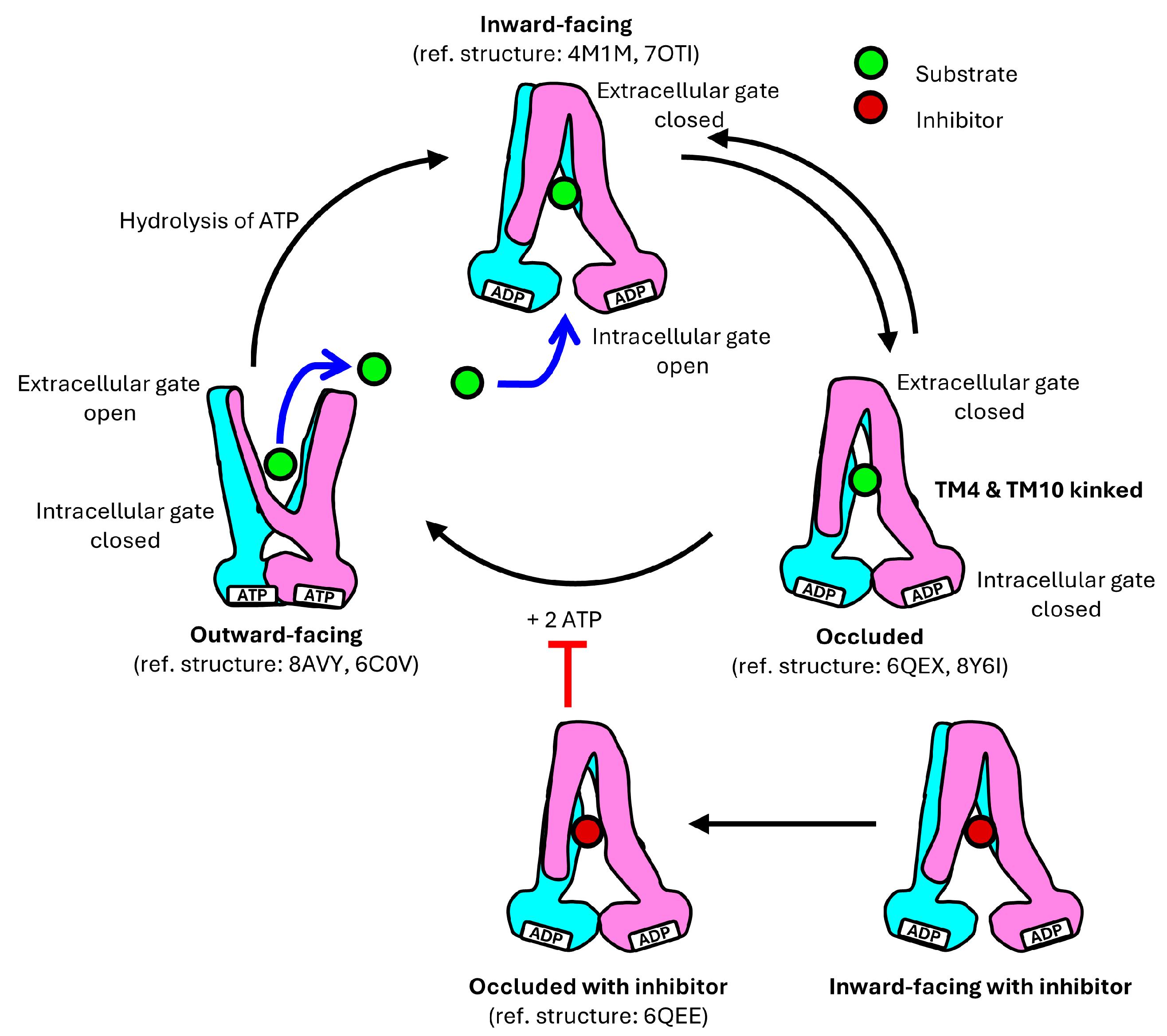
4. Translocation Mechanism of P-gp
5. Structural Insights into P-gp Mutants and Their Role in Multidrug Resistance
5.1. Summary of Key P-gp Mutants Associated with Multidrug Resistance in Chemotherapy
5.2. Analysis of Specific P-gp Mutants Related to Cancer Treatment Outcomes
5.3. Proposed Mechanisms of P-gp Mutants in Multidrug Resistance Based on Structural Insights
5.3.1. A61G (Asn21Asp)
5.3.2. G1199A (Ser400Asn)
5.3.3. T2677G/A (Ser893Ala/Thr)
5.3.4. T3421A (Ser1077Thr)
5.3.5. C1236T (Gly412Gly)
5.3.6. C3435T (Ile1145Ile)
6. Inhibitor Design and Potential Therapeutic Strategies
7. Areas for Further Research
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- American Cancer Society. Cancer Facts & Figures 2024; American Cancer Society: Atlanta, GA, USA, 2024. [Google Scholar]
- Gerlach, J.H.; Kartner, N.; Bell, D.R.; Ling, V. Multidrug resistance. Cancer Surv. 1986, 5, 25–46. [Google Scholar] [PubMed]
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel strategies to prevent the development of multidrug resistance (MDR) in cancer. Oncotarget 2017, 8, 84559–84571. [Google Scholar] [CrossRef] [PubMed]
- Catalano, A.; Iacopetta, D.; Ceramella, J.; Scumaci, D.; Giuzio, F.; Saturnino, C.; Aquaro, S.; Rosano, C.; Sinicropi, M.S. Multidrug Resistance (MDR): A Widespread Phenomenon in Pharmacological Therapies. Molecules 2022, 27, 616. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, H.; Assaraf, Y.G.; Zhao, K.; Xu, X.; Xie, J.; Yang, D.-H.; Chen, Z.-S. Overcoming ABC transporter-mediated multidrug resistance: Molecular mechanisms and novel therapeutic drug strategies. Drug Resist. Updates 2016, 27, 14–29. [Google Scholar] [CrossRef]
- Björn, N.; Jakobsen Falk, I.; Vergote, I.; Gréen, H. ABCB1 Variation Affects Myelosuppression, Progression-free Survival and Overall Survival in Paclitaxel/Carboplatin-treated Ovarian Cancer Patients. Basic Clin. Pharmacol. Toxicol. 2018, 123, 277–287. [Google Scholar] [CrossRef]
- Johnatty, S.E.; Beesley, J.; Paul, J.; Fereday, S.; Spurdle, A.B.; Webb, P.M.; Byth, K.; Marsh, S.; McLeod, H.; Group, A.S.; et al. ABCB1 (MDR 1) polymorphisms and progression-free survival among women with ovarian cancer following paclitaxel/carboplatin chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5594–5601. [Google Scholar] [CrossRef]
- Li, W.; Zhang, D.; Du, F.; Xing, X.; Wu, Y.; Xiao, D.; Liang, M.; Fan, Z.; Zhao, P.; Liu, T.; et al. ABCB1 3435TT and ABCG2 421CC genotypes were significantly associated with longer progression-free survival in Chinese breast cancer patients. Oncotarget 2017, 8, 111041–111052. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Thiebaut, F.; Tsuruo, T.; Hamada, H.; Gottesman, M.M.; Pastan, I.; Willingham, M.C. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 7735. [Google Scholar] [CrossRef]
- Schinkel, A.H.; Jonker, J.W. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv. Drug Del. Rev. 2003, 55, 3–29. [Google Scholar] [CrossRef]
- Staud, F.; Ceckova, M.; Micuda, S.; Pavek, P. Expression and Function of P-Glycoprotein in Normal Tissues: Effect on Pharmacokinetics. In Multi-Drug Resistance in Cancer; Zhou, J., Ed.; Humana Press: Totowa, NJ, USA, 2010; pp. 199–222. [Google Scholar]
- Engle, K.; Kumar, G. Cancer multidrug-resistance reversal by ABCB1 inhibition: A recent update. Eur. J. Med. Chem. 2022, 239, 114542. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Arias, I.M. Intracellular trafficking of P-glycoprotein. Int. J. Biochem. Cell Biol. 2012, 44, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Sajid, A.; Rahman, H.; Ambudkar, S.V. Advances in the structure, mechanism and targeting of chemoresistance-linked ABC transporters. Nat. Rev. Cancer 2023, 23, 762–779. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. Complex Interplay between the P-Glycoprotein Multidrug Efflux Pump and the Membrane: Its Role in Modulating Protein Function. Front. Oncol. 2014, 4, 41. [Google Scholar] [CrossRef]
- Syed, S.B.; Lin, S.-Y.; Arya, H.; Fu, I.H.; Yeh, T.-K.; Charles, M.R.C.; Periyasamy, L.; Hsieh, H.-P.; Coumar, M.S. Overcoming vincristine resistance in cancer: Computational design and discovery of piperine-inspired P-glycoprotein inhibitors. Chem. Biol. Drug Des. 2021, 97, 51–66. [Google Scholar] [CrossRef]
- Chufan, E.E.; Sim, H.-M.; Ambudkar, S.V. Molecular Basis of the Polyspecificity of P-Glycoprotein (ABCB1): Recent Biochemical and Structural Studies. Adv. Cancer Res. 2015, 125, 71–96. [Google Scholar]
- Nosol, K.; Romane, K.; Irobalieva, R.N.; Alam, A.; Kowal, J.; Fujita, N.; Locher, K.P. Cryo-EM structures reveal distinct mechanisms of inhibition of the human multidrug transporter ABCB1. Proc. Natl. Acad. Sci. USA 2020, 117, 26245–26253. [Google Scholar] [CrossRef]
- Thomas, C.; Tampé, R. Structural and Mechanistic Principles of ABC Transporters. Annu. Rev. Biochem. 2020, 89, 605–636. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef]
- Alam, A.; Küng, R.; Kowal, J.; McLeod, R.A.; Tremp, N.; Broude, E.V.; Roninson, I.B.; Stahlberg, H.; Locher, K.P. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. USA 2018, 115, E1973–E1982. [Google Scholar] [CrossRef]
- Hamaguchi-Suzuki, N.; Adachi, N.; Moriya, T.; Yasuda, S.; Kawasaki, M.; Suzuki, K.; Ogasawara, S.; Anzai, N.; Senda, T.; Murata, T. Cryo-EM structure of P-glycoprotein bound to triple elacridar inhibitor molecules. Biochem. Biophys. Res. Commun. 2024, 709, 149855. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef]
- Kurre, D.; Dang, P.X.; Le, L.T.M.; Gadkari, V.V.; Alam, A. Structural insights into binding-site access and ligand recognition by human ABCB1. EMBO J. 2025, 44, 991–1006. [Google Scholar] [CrossRef]
- Urgaonkar, S.; Nosol, K.; Said, A.M.; Nasief, N.N.; Bu, Y.; Locher, K.P.; Lau, J.Y.N.; Smolinski, M.P. Discovery and Characterization of Potent Dual P-Glycoprotein and CYP3A4 Inhibitors: Design, Synthesis, Cryo-EM Analysis, and Biological Evaluations. J. Med. Chem. 2022, 65, 191–216. [Google Scholar] [CrossRef]
- Gewering, T.; Waghray, D.; Parey, K.; Jung, H.; Tran, N.N.B.; Zapata, J.; Zhao, P.; Chen, H.; Januliene, D.; Hummer, G.; et al. Tracing the substrate translocation mechanism in P-glycoprotein. eLife 2024, 12, RP90174. [Google Scholar] [CrossRef]
- Barbieri, A.; Thonghin, N.; Shafi, T.; Prince, S.M.; Collins, R.F.; Ford, R.C. Structure of ABCB1/P-Glycoprotein in the Presence of the CFTR Potentiator Ivacaftor. Membranes 2021, 11, 923. [Google Scholar] [CrossRef]
- Le, C.A.; Harvey, D.S.; Aller, S.G. Structural definition of polyspecific compensatory ligand recognition by P-glycoprotein. IUCrJ 2020, 7, 663–672. [Google Scholar] [CrossRef]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Matsuoka, K.; Kimura, Y.; Ueda, K.; Kato, H. Inward- and outward-facing X-ray crystal structures of homodimeric P-glycoprotein CmABCB1. Nat. Commun. 2019, 10, 88. [Google Scholar] [CrossRef]
- Thonghin, N.; Collins, R.F.; Barbieri, A.; Shafi, T.; Siebert, A.; Ford, R.C. Novel features in the structure of P-glycoprotein (ABCB1) in the post-hydrolytic state as determined at 7.9 Å resolution. BMC Struct. Biol. 2018, 18, 17. [Google Scholar] [CrossRef]
- Esser, L.; Zhou, F.; Pluchino, K.M.; Shiloach, J.; Ma, J.; Tang, W.-k.; Gutierrez, C.; Zhang, A.; Shukla, S.; Madigan, J.P.; et al. Structures of the Multidrug Transporter P-glycoprotein Reveal Asymmetric ATP Binding and the Mechanism of Polyspecificity. J. Biol. Chem. 2017, 292, 446–461. [Google Scholar] [CrossRef]
- Nicklisch, S.C.T.; Rees, S.D.; McGrath, A.P.; Gökirmak, T.; Bonito, L.T.; Vermeer, L.M.; Cregger, C.; Loewen, G.; Sandin, S.; Chang, G.; et al. Global marine pollutants inhibit P-glycoprotein: Environmental levels, inhibitory effects, and cocrystal structure. Sci. Adv. 2016, 2, e1600001. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, P.; Tao, H.; McGrath, A.P.; Villaluz, M.; Rees, S.D.; Lee, S.C.; Doshi, R.; Urbatsch, I.L.; Zhang, Q.; Chang, G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. Sect. D Struct. Biol. 2015, 71, 732–741. [Google Scholar] [CrossRef] [PubMed]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Sakiyama, K.; Hipolito, C.J.; Fujioka, A.; Hirokane, R.; Ikeguchi, K.; Watanabe, B.; Hiratake, J.; et al. Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. USA 2014, 111, 4049–4054. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jaimes, K.F.; Aller, S.G. Refined structures of mouse P-glycoprotein. Protein Sci. 2014, 23, 34–46. [Google Scholar] [CrossRef]
- Ward, A.B.; Szewczyk, P.; Grimard, V.; Lee, C.-W.; Martinez, L.; Doshi, R.; Caya, A.; Villaluz, M.; Pardon, E.; Cregger, C.; et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef]
- Ambudkar, S.V.; Dey, S.; Hrycyna, C.A.; Ramachandra, M.; Pastan, I.; Gottesman, M.M. Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 361–398. [Google Scholar] [CrossRef]
- Rosenberg, M.F.; Callaghan, R.; Ford, R.C.; Higgins, C.F. Structure of the multidrug resistance P-glycoprotein to 2.5 nm resolution determined by electron microscopy and image analysis. J. Biol. Chem. 1997, 272, 10685–10694. [Google Scholar] [CrossRef]
- Huang, J.; Ecker, G.F. A Structure-Based View on ABC-Transporter Linked to Multidrug Resistance. Molecules 2023, 28, 495. [Google Scholar] [CrossRef]
- Elbahnsi, A.; Dudas, B.; Cisternino, S.; Declèves, X.; Miteva, M.A. Mechanistic insights into P-glycoprotein ligand transport and inhibition revealed by enhanced molecular dynamics simulations. Comput. Struct. Biotechnol. J. 2024, 23, 2548–2564. [Google Scholar] [CrossRef]
- Li, H.; Gong, W. Study of Allosteric Transitions of Human P-Glycoprotein by Using the Two-State Anisotropic Network Model. Front. Med. 2022, 9, 815355. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Kodan, A.; Kimura, Y.; Ueda, K.; Nakatsu, T.; Kato, H. Functional role of the linker region in purified human P-glycoprotein. FEBS J. 2009, 276, 3504–3516. [Google Scholar] [CrossRef] [PubMed]
- Hrycyna, C.A.; Airan, L.E.; Germann, U.A.; Ambudkar, S.V.; Pastan, I.; Gottesman, M.M. Structural flexibility of the linker region of human P-glycoprotein permits ATP hydrolysis and drug transport. Biochemistry 1998, 37, 13660–13673. [Google Scholar] [CrossRef] [PubMed]
- Nuti, S.L.; Rao, U.S. Proteolytic Cleavage of the Linker Region of the Human P-glycoprotein Modulates Its ATPase Function. J. Biol. Chem. 2002, 277, 29417–29423. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.-J.U.; dos Santos, D.J.V.A. Assessing the Stabilization of P-Glycoprotein’s Nucleotide-Binding Domains by the Linker, Using Molecular Dynamics. Mol. Inform. 2013, 32, 529–540. [Google Scholar] [CrossRef]
- Shapiro, A.B.; Ling, V. Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem. 1997, 250, 130–137. [Google Scholar] [CrossRef]
- Lagares, L.M.; Pérez-Castillo, Y.; Minovski, N.; Novič, M. Structure–Function Relationships in the Human P-Glycoprotein (ABCB1): Insights from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2021, 23, 362. [Google Scholar] [CrossRef]
- Horio, M.; Gottesman, M.M.; Pastan, I. ATP-dependent transport of vinblastine in vesicles from human multidrug-resistant cells. Proc. Natl. Acad. Sci. USA 1988, 85, 3580–3584. [Google Scholar] [CrossRef]
- Hrycyna, C.A.; Ramachandra, M.; Germann, U.A.; Cheng, P.W.; Pastan, I.; Gottesman, M.M. Both ATP sites of human P-glycoprotein are essential but not symmetric. Biochemistry 1999, 38, 13887–13899. [Google Scholar] [CrossRef]
- Urbatsch, I.L.; Beaudet, L.; Carrier, I.; Gros, P. Mutations in either nucleotide-binding site of P-glycoprotein (Mdr3) prevent vanadate trapping of nucleotide at both sites. Biochemistry 1998, 37, 4592–4602. [Google Scholar] [CrossRef]
- Sauna, Z.E.; Kim, I.-W.; Nandigama, K.; Kopp, S.; Chiba, P.; Ambudkar, S.V. Catalytic Cycle of ATP Hydrolysis by P-Glycoprotein: Evidence for Formation of the E·S Reaction Intermediate with ATP-γ-S, a Nonhydrolyzable Analogue of ATP. Biochemistry 2007, 46, 13787–13799. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. The “LSGGQ” motif in each nucleotide-binding domain of human P-glycoprotein is adjacent to the opposing walker A sequence. J. Biol. Chem. 2002, 277, 41303–41306. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E.; Saraste, M.; Runswick, M.J.; Gay, N.J. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Ambudkar, S.V.; Kim, I.-W.; Xia, D.; Sauna, Z.E. The A-loop, a novel conserved aromatic acid subdomain upstream of the Walker A motif in ABC transporters, is critical for ATP binding. FEBS Lett. 2006, 580, 1049–1055. [Google Scholar] [CrossRef]
- Hollenstein, K.; Dawson, R.J.P.; Locher, K.P. Structure and mechanism of ABC transporter proteins. Curr. Opin. Struct. Biol. 2007, 17, 412–418. [Google Scholar] [CrossRef]
- Juvale, I.I.A.; Hamid, A.A.A.; Halim, K.B.A.; Has, A.T.C. P-glycoprotein: New insights into structure, physiological function, regulation and alterations in disease. Heliyon 2022, 8, e09777. [Google Scholar] [CrossRef]
- Yang, R.; Hou, Y.-x.; Campbell, C.A.; Palaniyandi, K.; Zhao, Q.; Bordner, A.J.; Chang, X.-b. Glutamine residues in Q-loops of multidrug resistance protein MRP1 contribute to ATP binding via interaction with metal cofactor. Biochim. Biophys. Acta 2011, 1808, 1790–1796. [Google Scholar] [CrossRef]
- Jones, P.M.; George, A.M. A new structural model for P-glycoprotein. J. Membr. Biol. 1998, 166, 133–147. [Google Scholar] [CrossRef]
- Zaitseva, J.; Jenewein, S.; Jumpertz, T.; Holland, I.B.; Schmitt, L. H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB. EMBO J. 2005, 24, 1901–1910. [Google Scholar] [CrossRef]
- Emmert, D.; Campos, C.R.; Ward, D.; Lu, P.; Namanja, H.A.; Bohn, K.; Miller, D.S.; Sharom, F.J.; Chmielewski, J.; Hrycyna, C.A. Reversible Dimers of the Atypical Antipsychotic Quetiapine Inhibit P-Glycoprotein-Mediated Efflux in Vitro with Increased Binding Affinity and in Situ at the Blood-Brain Barrier. ACS Chem. Neurosci. 2014, 5, 305–317. [Google Scholar] [CrossRef]
- Jagodinsky, J.C.; Akgun, U. Characterizing the binding interactions between P-glycoprotein and eight known cardiovascular transport substrates. Pharmacol. Res. Perspect. 2015, 3, e00114. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.-W.; Booth-Genthe, C.; Ambudkar, S.V. Relationship between drugs and functional activity of various mammalian P-glycoproteins (ABCB1). Mini Rev. Med. Chem. 2008, 8, 193–200. [Google Scholar]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J. Biol. Chem. 2003, 278, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- Rahman, H.; Ware, M.J.; Sajid, A.; Lusvarghi, S.; Durell, S.R.; Ambudkar, S.V. Residues from Homologous Transmembrane Helices 4 and 10 Are Critical for P-Glycoprotein (ABCB1)-Mediated Drug Transport. Cancers 2023, 15, 3459. [Google Scholar] [CrossRef] [PubMed]
- Calinsky, R.; Levy, Y. Aromatic Residues in Proteins: Re-Evaluating the Geometry and Energetics of π–π, Cation–π, and CH–π Interactions. J. Phys. Chem. B 2024, 128, 8687–8700. [Google Scholar] [CrossRef]
- Pluchino, K.M.; Hall, M.D.; Moen, J.K.; Chufan, E.E.; Fetsch, P.A.; Shukla, S.; Gill, D.R.; Hyde, S.C.; Xia, D.; Ambudkar, S.V.; et al. Human-Mouse Chimeras with Normal Expression and Function Reveal That Major Domain Swapping Is Tolerated by P-Glycoprotein (ABCB1). Biochemistry 2016, 55, 1010–1023. [Google Scholar] [CrossRef]
- Pan, L.; Aller, S.G. Allosteric Role of Substrate Occupancy Toward the Alignment of P-glycoprotein Nucleotide Binding Domains. Sci. Rep. 2018, 8, 14643. [Google Scholar] [CrossRef]
- Vahedi, S.; Chufan, E.E.; Ambudkar, S.V. Global alteration of the drug-binding pocket of human P-glycoprotein (ABCB1) by substitution of fifteen conserved residues reveals a negative correlation between substrate size and transport efficiency. Biochem. Pharmacol. 2017, 143, 53–64. [Google Scholar] [CrossRef]
- Tombline, G.; Senior, A.E. The Occluded Nucleotide Conformation of P-Glycoprotein. J. Bioenerg. Biomembr. 2005, 37, 497–500. [Google Scholar] [CrossRef]
- Siarheyeva, A.; Liu, R.; Sharom, F.J. Characterization of an Asymmetric Occluded State of P-glycoprotein with Two Bound Nucleotides. J. Biol. Chem. 2010, 285, 7575–7586. [Google Scholar] [CrossRef]
- Crowley, E.; O’Mara, M.L.; Kerr, I.D.; Callaghan, R. Transmembrane helix 12 plays a pivotal role in coupling energy provision and drug binding in ABCB1. FEBS J. 2010, 277, 3974–3985. [Google Scholar] [CrossRef] [PubMed]
- Crowley, E.; O’Mara, M.L.; Reynolds, C.; Tieleman, D.P.; Storm, J.; Kerr, I.D.; Callaghan, R. Transmembrane Helix 12 Modulates Progression of the ATP Catalytic Cycle in ABCB1. Biochemistry 2009, 48, 6249–6258. [Google Scholar] [CrossRef] [PubMed]
- Kimchi-Sarfaty, C.; Gribar, J.J.; Gottesman, M.M. Functional characterization of coding polymorphisms in the human MDR1 gene using a vaccinia virus expression system. Mol. Pharmacol. 2002, 62, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Tennakoon, L.; Keller, J.; Sarginson, J.E.; Ryan, H.S.; Murphy, G.M.; Lazzeroni, L.C.; Trivedi, M.H.; Kocsis, J.H.; DeBattista, C.; et al. ABCB1 (MDR1) predicts remission on P-gp substrates in chronic depression. Pharmacogenom. J. 2015, 15, 332–339. [Google Scholar] [CrossRef]
- Ruiz, J.; Herrero, M.J.; Bosó, V.; Megías, J.E.; Hervás, D.; Poveda, J.L.; Escrivá, J.; Pastor, A.; Solé, A.; Aliño, S.F. Impact of Single Nucleotide Polymorphisms (SNPs) on Immunosuppressive Therapy in Lung Transplantation. Int. J. Mol. Sci. 2015, 16, 20168–20182. [Google Scholar] [CrossRef]
- Woodahl, E.L.; Yang, Z.; Bui, T.; Shen, D.D.; Ho, R.J.Y. Multidrug Resistance Gene G1199A Polymorphism Alters Efflux Transport Activity of P-Glycoprotein. J. Pharmacol. Exp. Ther. 2004, 310, 1199–1207. [Google Scholar] [CrossRef]
- Crouthamel, M.H.; Wu, D.; Yang, Z.; Ho, R.J.Y. A Novel MDR1 G1199T Variant Alters Drug Resistance and Efflux Transport Activity of P-Glycoprotein in Recombinant Hek Cells. J. Pharm. Sci. 2006, 95, 2767–2777. [Google Scholar] [CrossRef]
- Woodahl, E.L.; Yang, Z.; Bui, T.; Shen, D.D.; Ho, R.J. MDR1 G1199A polymorphism alters permeability of HIV protease inhibitors across P-glycoprotein-expressing epithelial cells. AIDS 2005, 19, 1617–1625. [Google Scholar] [CrossRef]
- Tecza, K.; Pamula-Pilat, J.; Lanuszewska, J.; Grzybowska, E. Genetic polymorphisms and response to 5-fluorouracil, doxorubicin and cyclophosphamide chemotherapy in breast cancer patients. Oncotarget 2016, 7, 66790. [Google Scholar] [CrossRef]
- Kim, J.-H.; Kim, S.R.; Song, I.-S.; Shin, H.-J.; Kim, H.-S.; Lee, J.-H.; Ko, S.-G.; Shin, Y.-C. Different transport activity of human triallelic MDR1 893Ala/Ser/Thr variant and its association with herb extracts. Phytother. Res. PTR 2011, 25, 1141–1147. [Google Scholar] [CrossRef]
- Lewandrowski, K.-U.; Sharafshah, A.; Elfar, J.; Schmidt, S.L.; Blum, K.; Wetzel, F.T. A Pharmacogenomics-Based In Silico Investigation of Opioid Prescribing in Post-operative Spine Pain Management and Personalized Therapy. Cell. Mol. Neurobiol. 2024, 44, 47. [Google Scholar] [CrossRef] [PubMed]
- Hemauer, S.J.; Nanovskaya, T.N.; Abdel-Rahman, S.Z.; Patrikeeva, S.L.; Hankins, G.D.V.; Ahmed, M.S. Modulation of human placental P-glycoprotein expression and activity by MDR1 gene polymorphisms. Biochem. Pharmacol. 2010, 79, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.B.; Leake, B.F.; Choo, E.F.; Dresser, G.K.; Kubba, S.V.; Schwarz, U.I.; Taylor, A.; Xie, H.-G.; McKinsey, J.; Zhou, S.; et al. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin. Pharmacol. Ther. 2001, 70, 189–199. [Google Scholar] [CrossRef]
- Leschziner, G.; Zabaneh, D.; Pirmohamed, M.; Owen, A.; Rogers, J.; Coffey, A.J.; Balding, D.J.; Bentley, D.B.; Johnson, M.R. Exon sequencing and high resolution haplotype analysis of ABC transporter genes implicated in drug resistance. Pharmacogenet. Genom. 2006, 16, 439. [Google Scholar] [CrossRef]
- Tang, K.; Ngoi, S.-M.; Gwee, P.-C.; Chua, J.M.Z.; Lee, E.J.D.; Chong, S.S.; Lee, C.G.L. Distinct haplotype profiles and strong linkage disequilibrium at the MDR1 multidrug transporter gene locus in three ethnic Asian populations. Pharmacogenetics 2002, 12, 437–450. [Google Scholar] [CrossRef]
- Dizdarevic, S.; Peters, A.M. Imaging of multidrug resistance in cancer. Cancer Imaging 2011, 11, 1–8. [Google Scholar] [CrossRef]
- Hitzl, M.; Drescher, S.; van der Kuip, H.; Schäffeler, E.; Fischer, J.; Schwab, M.; Eichelbaum, M.; Fromm, M.F. The C3435T mutation in the human MDR1 gene is associated with altered efflux of the P-glycoprotein substrate rhodamine 123 from CD56+ natural killer cells. Pharmacogenetics 2001, 11, 293–298. [Google Scholar] [CrossRef]
- Hoffmeyer, S.; Burk, O.; von Richter, O.; Arnold, H.P.; Brockmöller, J.; Johne, A.; Cascorbi, I.; Gerloff, T.; Roots, I.; Eichelbaum, M.; et al. Functional polymorphisms of the human multidrug-resistance gene: Multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 3473–3478. [Google Scholar] [CrossRef]
- Johne, A.; Köpke, K.; Gerloff, T.; Mai, I.; Rietbrock, S.; Meisel, C.; Hoffmeyer, S.; Kerb, R.; Fromm, M.F.; Brinkmann, U.; et al. Modulation of steady-state kinetics of digoxin by haplotypes of the P-glycoprotein MDR1 gene. Clin. Pharmacol. Ther. 2002, 72, 584–594. [Google Scholar] [CrossRef]
- Kurata, Y.; Ieiri, I.; Kimura, M.; Morita, T.; Irie, S.; Urae, A.; Ohdo, S.; Ohtani, H.; Sawada, Y.; Higuchi, S.; et al. Role of human MDR1 gene polymorphism in bioavailability and interaction of digoxin, a substrate of P-glycoprotein. Clin. Pharmacol. Ther. 2002, 72, 209–219. [Google Scholar] [CrossRef]
- Wang, D.; Johnson, A.D.; Papp, A.C.; Kroetz, D.L.; Sadée, W. Multidrug resistance polypeptide 1 (MDR1, ABCB1) variant 3435C>T affects mRNA stability. Pharmacogenet. Genom. 2005, 15, 693–704. [Google Scholar] [CrossRef]
- Fung, K.L.; Gottesman, M.M. A synonymous polymorphism in a common MDR1 (ABCB1) haplotype shapes protein function. Biochim. Biophys. Acta 2009, 1794, 860–871. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-J.; Sauna, Z.E.; Kimchi-Sarfaty, C.; Ambudkar, S.V.; Gottesman, M.M.; Nussinov, R. Synonymous mutations and ribosome stalling can lead to altered folding pathways and distinct minima. J. Mol. Biol. 2008, 383, 281–291. [Google Scholar] [CrossRef]
- Jeong, S.-H.; Jang, J.-H.; Lee, Y.-B. P-glycoprotein mechanical functional analysis using in silico molecular modeling: Pharmacokinetic variability according to ABCB1 c.2677G>T/A genetic polymorphisms. Int. J. Biol. Macromol. 2023, 249, 126777. [Google Scholar] [CrossRef]
- Kimchi-Sarfaty, C.; Oh, J.M.; Kim, I.-W.; Sauna, Z.E.; Calcagno, A.M.; Ambudkar, S.V.; Gottesman, M.M. A “Silent” Polymorphism in the MDR1 Gene Changes Substrate Specificity. Science 2007, 315, 525–528. [Google Scholar] [CrossRef]
- Subramanian, K.; Payne, B.; Feyertag, F.; Alvarez-Ponce, D. The Codon Statistics Database: A Database of Codon Usage Bias. Mol. Biol. Evol. 2022, 39, msac157. [Google Scholar] [CrossRef]
- Adzhubei, A.A.; Adzhubeib, I.A.; Krasheninnikov, I.A.; Neidle, S. Non-random usage of ‘degenerate’ codons is related to protein three-dimensional structure. FEBS Lett. 1996, 399, 78–82. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Q.; Zhao, F. Synonymous but not Silent: The Codon Usage Code for Gene Expression and Protein Folding. Annu. Rev. Biochem. 2021, 90, 375–401. [Google Scholar] [CrossRef]
- Xie, T.; Ding, D. The relationship between synonymous codon usage and protein structure. FEBS Lett. 1998, 434, 93–96. [Google Scholar]
- Brant, S.R.; Panhuysen, C.I.M.; Nicolae, D.; Reddy, D.M.; Bonen, D.K.; Karaliukas, R.; Zhang, L.; Swanson, E.; Datta, L.W.; Moran, T.; et al. MDR1 Ala893 Polymorphism Is Associated with Inflammatory Bowel Disease. Am. J. Hum. Genet. 2003, 73, 1282–1292. [Google Scholar] [CrossRef]
- Gao, Y.; Wei, C.; Luo, L.; Tang, Y.; Yu, Y.; Li, Y.; Xing, J.; Pan, X. Membrane-assisted tariquidar access and binding mechanisms of human ATP-binding cassette transporter P-glycoprotein. Front. Mol. Biosci. 2024, 11, 1364494. [Google Scholar] [CrossRef] [PubMed]
- Salazar, P.B.; Murakami, M.; Ranganathan, N.; Durell, S.R.; Ambudkar, S.V. Mutational analysis reveals the importance of residues of the access tunnel inhibitor site to human P-glycoprotein (ABCB1)-mediated transport. Protein Sci. Publ. Protein Soc. 2024, 33, e5155. [Google Scholar] [CrossRef] [PubMed]
- Bankstahl, J.P.; Bankstahl, M.; Römermann, K.; Wanek, T.; Stanek, J.; Windhorst, A.D.; Fedrowitz, M.; Erker, T.; Müller, M.; Löscher, W.; et al. Tariquidar and elacridar are dose-dependently transported by P-glycoprotein and Bcrp at the blood-brain barrier: A small-animal positron emission tomography and in vitro study. Drug Metab. Dispos. 2013, 41, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Galetin, A.; Brouwer, K.L.R.; Tweedie, D.; Yoshida, K.; Sjöstedt, N.; Aleksunes, L.; Chu, X.; Evers, R.; Hafey, M.J.; Lai, Y.; et al. Membrane transporters in drug development and as determinants of precision medicine. Nat. Rev. Drug Discov. 2024, 23, 255–280. [Google Scholar] [CrossRef]
- Lei, Z.-N.; Teng, Q.-X.; Wu, Z.-X.; Ping, F.-F.; Song, P.; Wurpel, J.N.D.; Chen, Z.-S. Overcoming multidrug resistance by knockout of ABCB1 gene using CRISPR/Cas9 system in SW620/Ad300 colorectal cancer cells. MedComm 2021, 2, 765–777. [Google Scholar] [CrossRef]
- Fleming, T.J.; Schrankel, C.S.; Vyas, H.; Rosenblatt, H.D.; Hamdoun, A. CRISPR/Cas9 mutagenesis reveals a role for ABCB1 in gut immune responses to Vibrio diazotrophicus in sea urchin larvae. J. Exp. Biol. 2021, 224, jeb232272. [Google Scholar] [CrossRef]
- Yang, Y.; Qiu, J.-G.; Li, Y.; Di, J.-M.; Zhang, W.-J.; Jiang, Q.-W.; Zheng, D.-W.; Chen, Y.; Wei, M.-N.; Huang, J.-R.; et al. Targeting ABCB1-mediated tumor multidrug resistance by CRISPR/Cas9-based genome editing. Am. J. Transl. Res. 2016, 8, 3986–3994. [Google Scholar]
- Megías-Vericat, J.E.; Rojas, L.; Herrero, M.J.; Bosó, V.; Montesinos, P.; Moscardó, F.; Poveda, J.L.; Sanz, M.Á.; Aliño, S.F. Influence of ABCB1 polymorphisms upon the effectiveness of standard treatment for acute myeloid leukemia: A systematic review and meta-analysis of observational studies. Pharmacogenom. J. 2015, 15, 109–118. [Google Scholar] [CrossRef]
- Jiang, Q.; Xu, M.; Liu, Y.; Chen, Y.; Feng, J.; Wang, X.; Liang, S.; Li, D.; Yang, X. Influence of the ABCB1 polymorphisms on the response to Taxane-containing chemotherapy: A systematic review and meta-analysis. Cancer Chemother. Pharmacol. 2018, 81, 315–323. [Google Scholar] [CrossRef]
- Sun, S.; Cai, J.; Yang, Q.; Zhu, Y.; Zhao, S.; Wang, Z. Prognostic Value and Implication for Chemotherapy Treatment of ABCB1 in Epithelial Ovarian Cancer: A Meta-Analysis. PLoS ONE 2016, 11, e0166058. [Google Scholar] [CrossRef]
- Pigott, T.D.; Polanin, J.R. Methodological Guidance Paper: High-Quality Meta-Analysis in a Systematic Review. Rev. Educ. Res. 2020, 90, 24–46. [Google Scholar] [CrossRef]
- Skinner, K.T.; Palkar, A.M.; Hong, A.L. Genetics of ABCB1 in Cancer. Cancers 2023, 15, 4236. [Google Scholar] [CrossRef] [PubMed]
- Chung, F.S.; Santiago, J.S.; Jesus, M.F.M.D.; Trinidad, C.V.; See, M.F.E. Disrupting P-glycoprotein function in clinical settings: What can we learn from the fundamental aspects of this transporter? Am. J. Cancer Res. 2016, 6, 1583. [Google Scholar] [PubMed]
- Ogihara, T.; Kamiya, M.; Ozawa, M.; Fujita, T.; Yamamoto, A.; Yamashita, S.; Ohnishi, S.; Isomura, Y. What kinds of substrates show P-glycoprotein-dependent intestinal absorption? Comparison of verapamil with vinblastine. Drug Metab. Pharmacokinet. 2006, 21, 238–244. [Google Scholar] [CrossRef]
- Nervi, P.; Li-Blatter, X.; Aänismaa, P.; Seelig, A. P-glycoprotein substrate transport assessed by comparing cellular and vesicular ATPase activity. Biochim. Biophys. Acta 2010, 1798, 515–525. [Google Scholar] [CrossRef]
- Findlay, J.M.; Middleton, M.R.; Tomlinson, I. A systematic review and meta-analysis of somatic and germline DNA sequence biomarkers of esophageal cancer survival, therapy response and stage. Ann. Oncol. 2015, 26, 624–644. [Google Scholar] [CrossRef]
- Kaya, P.; Gündüz, U.; Arpaci, F.; Ural, A.U.; Guran, S. Identification of polymorphisms on the MDR1 gene among Turkish population and their effects on multidrug resistance in acute leukemia patients. Am. J. Hematol. 2005, 80, 26–34. [Google Scholar] [CrossRef]
- Peethambaram, P.; Fridley, B.L.; Vierkant, R.A.; Larson, M.C.; Kalli, K.R.; Elliott, E.A.; Oberg, A.L.; White, K.L.; Rider, D.N.; Keeney, G.L.; et al. Polymorphisms in ABCB1 and ERCC2 associated with ovarian cancer outcome. Int. J. Mol. Epidemiol. Genet 2011, 2, 185–195. [Google Scholar]
- Johnatty, S.E.; Beesley, J.; Gao, B.; Chen, X.; Lu, Y.; Law, M.H.; Henderson, M.J.; Russell, A.J.; Hedditch, E.L.; Emmanuel, C.; et al. ABCB1 (MDR1) polymorphisms and ovarian cancer progression and survival: A comprehensive analysis from the Ovarian Cancer Association Consortium and The Cancer Genome Atlas. Gynecol. Oncol. 2013, 131, 8–14. [Google Scholar] [CrossRef]
- Madejczyk, A.M.; Canzian, F.; Góra-Tybor, J.; Campa, D.; Sacha, T.; Link-Lenczowska, D.; Florek, I.; Prejzner, W.; Całbecka, M.; Rymko, M.; et al. Impact of genetic polymorphisms of drug transporters ABCB1 and ABCG2 and regulators of xenobiotic transport and metabolism PXR and CAR on clinical efficacy of dasatinib in chronic myeloid leukemia. Front. Oncol. 2022, 12, 952640. [Google Scholar] [CrossRef]
- Herrero Rivera, D.; Vacas, C.G.; Kovandzic, L.M.; Vázquez, J.P.; Alonso, L.A.; González, B.M.; Aragón, V.C.; Grande, E.; Caro, R.L.; Virizuela Echaburu, J.A.; et al. Single-Nucleotide Polymorphism Associations with Efficacy and Toxicity in Metastatic Castration-Resistant Prostate Cancer Treated with Cabazitaxel. Pharmacogenomics 2022, 23, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Gréen, H.; Falk, I.J.; Lotfi, K.; Paul, E.; Hermansson, M.; Rosenquist, R.; Paul, C.; Nahi, H. Association of ABCB1 polymorphisms with survival and in vitro cytotoxicty in de novo acute myeloid leukemia with normal karyotype. Pharmacogenom. J. 2012, 12, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Monzo, M.; Brunet, S.; Urbano-Ispizua, A.; Navarro, A.; Perea, G.; Esteve, J.; Artells, R.; Granell, M.; Berlanga, J.; Ribera, J.M.; et al. Genomic polymorphisms provide prognostic information in intermediate-risk acute myeloblastic leukemia. Blood 2006, 107, 4871–4879. [Google Scholar] [CrossRef]
- Rafiee, R.; Chauhan, L.; Alonzo, T.A.; Wang, Y.-C.; Elmasry, A.; Loken, M.R.; Pollard, J.; Aplenc, R.; Raimondi, S.; Hirsch, B.A.; et al. ABCB1 SNP predicts outcome in patients with acute myeloid leukemia treated with Gemtuzumab ozogamicin: A report from Children’s Oncology Group AAML0531 Trial. Blood Cancer J. 2019, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Macauda, A.; Castelli, E.; Buda, G.; Pelosini, M.; Butrym, A.; Watek, M.; Kruszewski, M.; Vangsted, A.J.; Rymko, M.; Jamroziak, K.; et al. Inherited variation in the xenobiotic transporter pathway and survival of multiple myeloma patients. Br. J. Haematol. 2018, 183, 375–384. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Organism | Mutant | Resolution (Å) | Conformation | Ligand | Nucleotide | Method | Year | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| 9CR8 | Homo sapiens | 3.80 | Inward-facing | cryo-EM | 2025 | [25] | |||
| 9CTC | Homo sapiens | 3.60 | Occluded | Zosuquidar | ATP | cryo-EM | 2025 | [25] | |
| 9CTF | Homo sapiens | 3.90 | Inward-facing | Taxol | ATP | cryo-EM | 2025 | [25] | |
| 9CTG | Homo sapiens | 3.40 | Outward-facing | ATPγS | cryo-EM | 2025 | [25] | ||
| 8Y6I | Homo sapiens, Mus musculus | 2.54 | Occluded | UIC2 Fab, elacridar | cryo-EM | 2024 | [23] | ||
| 8Y6H | Homo sapiens, Mus musculus | 2.49 | Occluded | UIC2 Fab, elacridar | cryo-EM | 2024 | [23] | ||
| 8PEE | Mus musculus | L335C | 3.8 | Inward-facing | AAC | cryo-EM | 2024 | [27] | |
| 8AVY | Mus musculus | L335C | 2.9 | Outward-facing | ATP | cryo-EM | 2023 | [27] | |
| 7ZKB | Mus musculus | V978C | 4.7 | Inward-facing | AAC | cryo-EM | 2023 | [27] | |
| 7ZKA | Mus musculus | V978C | 2.9 | Outward-facing | AAC | ATP | cryo-EM | 2023 | [27] |
| 7ZK9 | Mus musculus | L971C | 4.3 | Inward-facing | AAC | cryo-EM | 2023 | [27] | |
| 7ZK8 | Mus musculus | L971C | 3 | Outward-facing | AAC | ATP | cryo-EM | 2023 | [27] |
| 7ZK6 | Mus musculus | L335C | 3.1 | Outward-facing | AAC | ATP | cryo-EM | 2023 | [27] |
| 7ZK5 | Mus musculus | L335C | 2.6 | Outward-facing | AAC | ATP | cryo-EM | 2023 | [27] |
| 7ZK4 | Mus musculus | L335C | 2.6 | Outward-facing | ATP | cryo-EM | 2023 | [27] | |
| 7O9W | Homo sapiens, Mus musculus | 3.5 | Occluded | UIC2 Fab, encequidar | cryo-EM | 2022 | [26] | ||
| 7OTI | Mus musculus | 4.2 | Inward-facing | cryo-EM | 2021 | [28] | |||
| 7OTG | Mus musculus | 5.4 | Inward-facing | Ivacaftor | cryo-EM | 2021 | [28] | ||
| 7A69 | Homo sapiens | 3.2 | Occluded | MRK16 Fab, vincristine | cryo-EM | 2020 | [19] | ||
| 7A6F | Homo sapiens | 3.5 | Occluded | MRK16 Fab, zosuquidar | cryo-EM | 2020 | [19] | ||
| 7A6E | Homo sapiens | 3.6 | Occluded | MRK16 Fab, tariquidar | cryo-EM | 2020 | [19] | ||
| 7A6C | Homo sapiens | 3.6 | Occluded | MRK16 Fab, elacridar | cryo-EM | 2020 | [19] | ||
| 7A65 | Homo sapiens | 3.9 | Occluded | MRK16 Fab | cryo-EM | 2020 | [19] | ||
| 6UJW | Mus musculus | Y306A, C952A | 4.15 | Inward-facing | BDE100 | X-ray diffraction | 2020 | [29] | |
| 6UJT | Mus musculus | Y303A, C952A | 4.17 | Inward-facing | BDE100 | X-ray diffraction | 2020 | [29] | |
| 6UJS | Mus musculus | F728A, C952A | 4.17 | Inward-facing | BDE100 | X-ray diffraction | 2020 | [29] | |
| 6UJR | Mus musculus | F724A, C952A | 4.1 | Inward-facing | BDE100 | X-ray diffraction | 2020 | [29] | |
| 6UJP | Mus musculus | F979A, C952A | 3.98 | Inward-facing | BDE100 | X-ray diffraction | 2020 | [29] | |
| 6UJN | Mus musculus | C952A | 3.98 | Inward-facing | BDE100 | X-ray diffraction | 2020 | [29] | |
| 6QEX | Homo sapiens, Mus musculus | 3.6 | Occluded | UIC2 Fab, taxol | cryo-EM | 2019 | [21] | ||
| 6QEE | Homo sapiens, Mus musculus | 3.9 | Occluded | UIC2 Fab, zosuquidar | cryo-EM | 2019 | [21] | ||
| 6A6M | Cyanidioschyzon merolae | Q147A/T381A | 1.9 | Outward-facing | AMP-PNP | X-ray diffraction | 2019 | [30] | |
| 6A6N | Cyanidioschyzon merolae | Q147A/T381A | 3.02 | Inward-facing | X-ray diffraction | 2019 | [30] | ||
| 6Q81 | Mus musculus | 7.9 | Inward-facing | ADP | cryo-EM | 2018 | [31] | ||
| 6GDI | Mus musculus | 7.9 | Inward-facing | cryo-EM | 2018 | [31] | |||
| 6FN4 | Homo sapiens, Mus musculus | S559C, S1204C | 4.14 | Occluded | UIC2 Fab | cryo-EM | 2018 | [22] | |
| 6FN1 | Homo sapiens, Mus musculus | S559C, S1204C | 3.58 | Occluded | UIC2 Fab, zosuquidar | cryo-EM | 2018 | [22] | |
| 6C0V | Homo sapiens | E556Q, E1201Q | 3.4 | Outward-facing | ATP | cryo-EM | 2018 | [24] | |
| 5KOY | Mus musculus | Δ649–682 (34 linker deleted) | 3.85 | Inward-facing | ATP | X-ray diffraction | 2016 | [32] | |
| 5KPJ | Mus musculus | Methylated | 3.5 | Inward-facing | X-ray diffraction | 2016 | [32] | ||
| 5KPI | Mus musculus | 4.01 | Inward-facing | X-ray diffraction | 2016 | [32] | |||
| 5KPD | Mus musculus | E552Q, E1197Q, 34 linker deleted | 3.35 | Inward-facing | X-ray diffraction | 2016 | [32] | ||
| 5KO2 | Mus musculus | E552Q, E1197Q, 34 linker deleted | 3.3 | Inward-facing | Hg2+ | X-ray diffraction | 2016 | [32] | |
| 4XWK | Mus musculus | 3.5 | Inward-facing | BDE100 | X-ray diffraction | 2016 | [33] | ||
| 4Q9L | Mus musculus | 3.8 | Inward-facing | QZ-Phe | X-ray diffraction | 2015 | [34] | ||
| 4Q9K | Mus musculus | 3.8 | Inward-facing | QZ-Leu | X-ray diffraction | 2015 | [34] | ||
| 4Q9J | Mus musculus | 3.6 | Inward-facing | QZ-Val | X-ray diffraction | 2015 | [34] | ||
| 4Q9I | Mus musculus | 3.781 | Inward-facing | QZ-Ala | X-ray diffraction | 2015 | [34] | ||
| 4Q9H | Mus musculus | 3.4 | Inward-facing | X-ray diffraction | 2015 | [34] | |||
| 3WMG | Cyanidioschyzon merolae | G277V, A278V, A279V | 2.4 | Inward-facing | aCAP | X-ray diffraction | 2014 | [35] | |
| 3WMF | Cyanidioschyzon merolae | G277V, A278V, A279V | 2.6 | Inward-facing | X-ray diffraction | 2014 | [35] | ||
| 3WME | Cyanidioschyzon merolae | 2.751 | Inward-facing | X-ray diffraction | 2014 | [35] | |||
| 4M2T | Mus musculus | 4.35 | Inward-facing | QZ59-SSS | X-ray diffraction | 2013 | [36] | ||
| 4M2S | Mus musculus | 4.4 | Inward-facing | QZ59-RRR | X-ray diffraction | 2013 | [36] | ||
| 4M1M | Mus musculus | 3.8 | Inward-facing | X-ray diffraction | 2013 | [36] | |||
| 4KSD | Lama glama, Mus musculus | 4.1 | Inward-facing | NB592 | X-ray diffraction | 2013 | [37] | ||
| 4KSC | Mus musculus | 4 | Inward-facing | X-ray diffraction | 2013 | [37] | |||
| 4KSB | Mus musculus | 3.8001 | Inward-facing | X-ray diffraction | 2013 | [37] | |||
| 3G61 | Mus musculus | 4.35 | Inward-facing | QZ59-SSS | X-ray diffraction | 2009 | [38] | ||
| 3G60 | Mus musculus | 4.4 | Inward-facing | QZ59-RRR | X-ray diffraction | 2009 | [38] | ||
| 3G5U | Mus musculus | 3.8 | Inward-facing | X-ray diffraction | 2009 | [38] |
| SNP ID | Nucleotide Change | Amino Acid Change | Amino Acid | Mutation Type | Location | Phenotypic Effect and Clinical Implication |
|---|---|---|---|---|---|---|
| rs9282564 | A61G | Asn → Asp | 21 | Missense | Cytoplasmic loop before TM1 |
|
| rs2229109 | G1199A | Ser → Asn | 400 | Missense | NBD1 | |
| rs2032582 | T2677G/A | Ser → Ala/Thr | 893 | Missense | Cytoplasmic region of TM10 | |
| rs2229107 | T3421A | Ser → Thr | 1077 | Missense | NBD2 walker A motif |
|
| rs1128503 | C1236T | Gly → Gly | 412 | Silent | NBD1 | |
| rs1045642 | C3435T | Ile → Ile | 1145 | Silent | NBD2 |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tia, S.T.; Luo, M.; Fan, W. Mapping the Role of P-gp in Multidrug Resistance: Insights from Recent Structural Studies. Int. J. Mol. Sci. 2025, 26, 4179. https://doi.org/10.3390/ijms26094179
Tia ST, Luo M, Fan W. Mapping the Role of P-gp in Multidrug Resistance: Insights from Recent Structural Studies. International Journal of Molecular Sciences. 2025; 26(9):4179. https://doi.org/10.3390/ijms26094179
Chicago/Turabian StyleTia, Shi Ting, Min Luo, and Wenjie Fan. 2025. "Mapping the Role of P-gp in Multidrug Resistance: Insights from Recent Structural Studies" International Journal of Molecular Sciences 26, no. 9: 4179. https://doi.org/10.3390/ijms26094179
APA StyleTia, S. T., Luo, M., & Fan, W. (2025). Mapping the Role of P-gp in Multidrug Resistance: Insights from Recent Structural Studies. International Journal of Molecular Sciences, 26(9), 4179. https://doi.org/10.3390/ijms26094179