
The Binding of Brazilin from C. sappan to the Full-Length SARS-CoV-2 Spike Proteins

 , , and
, , and 
Abstract
1. Introduction
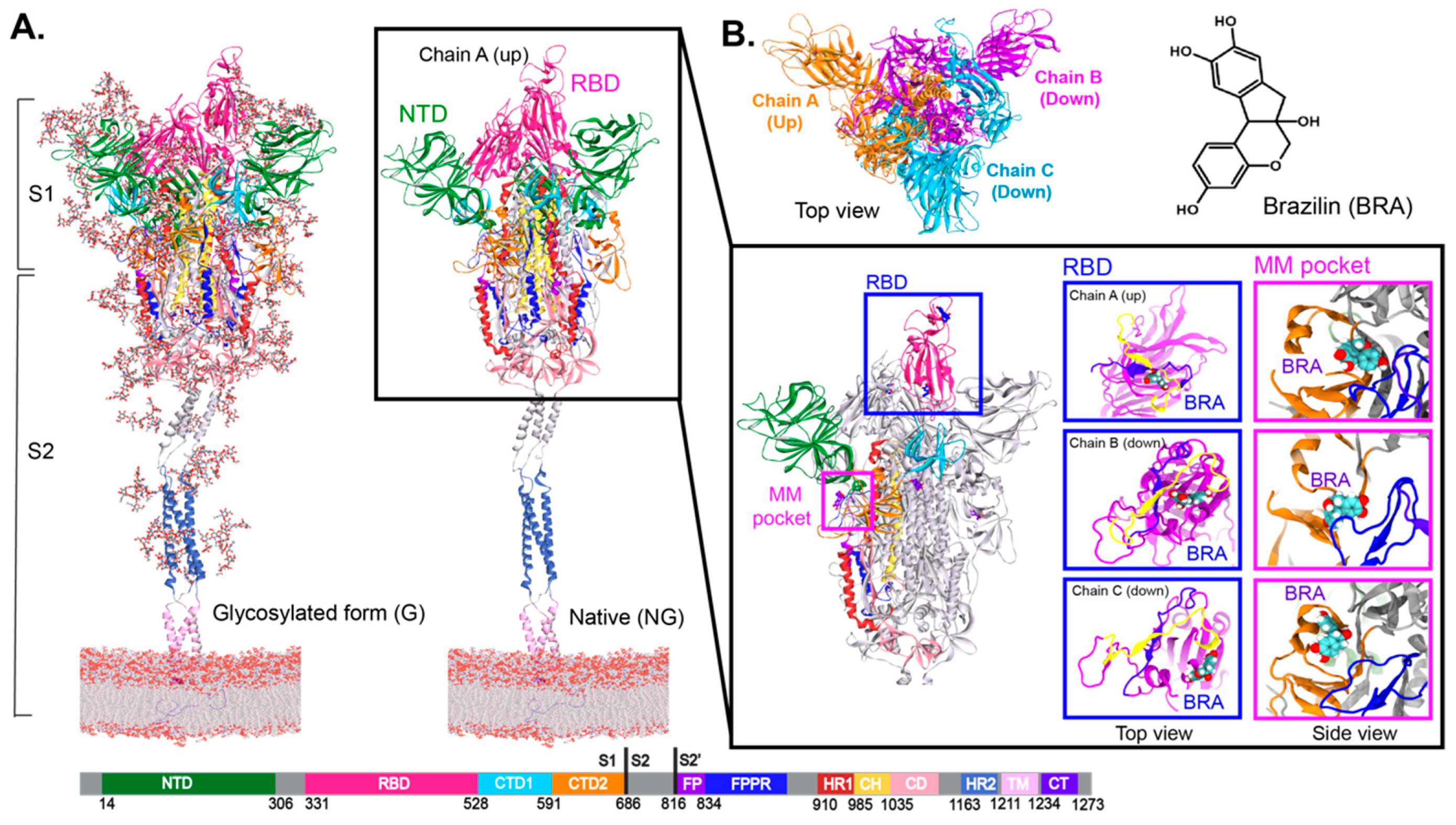
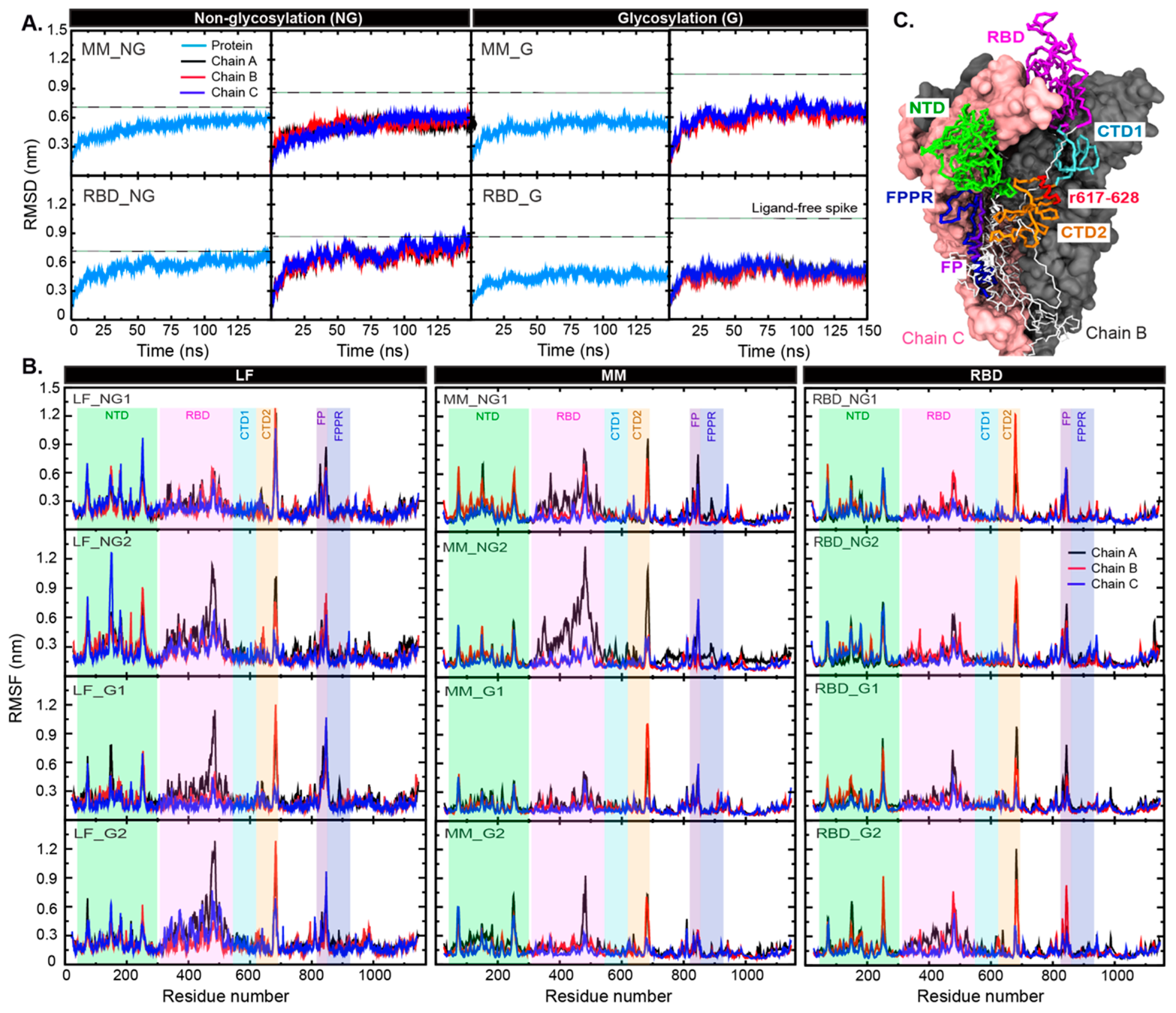
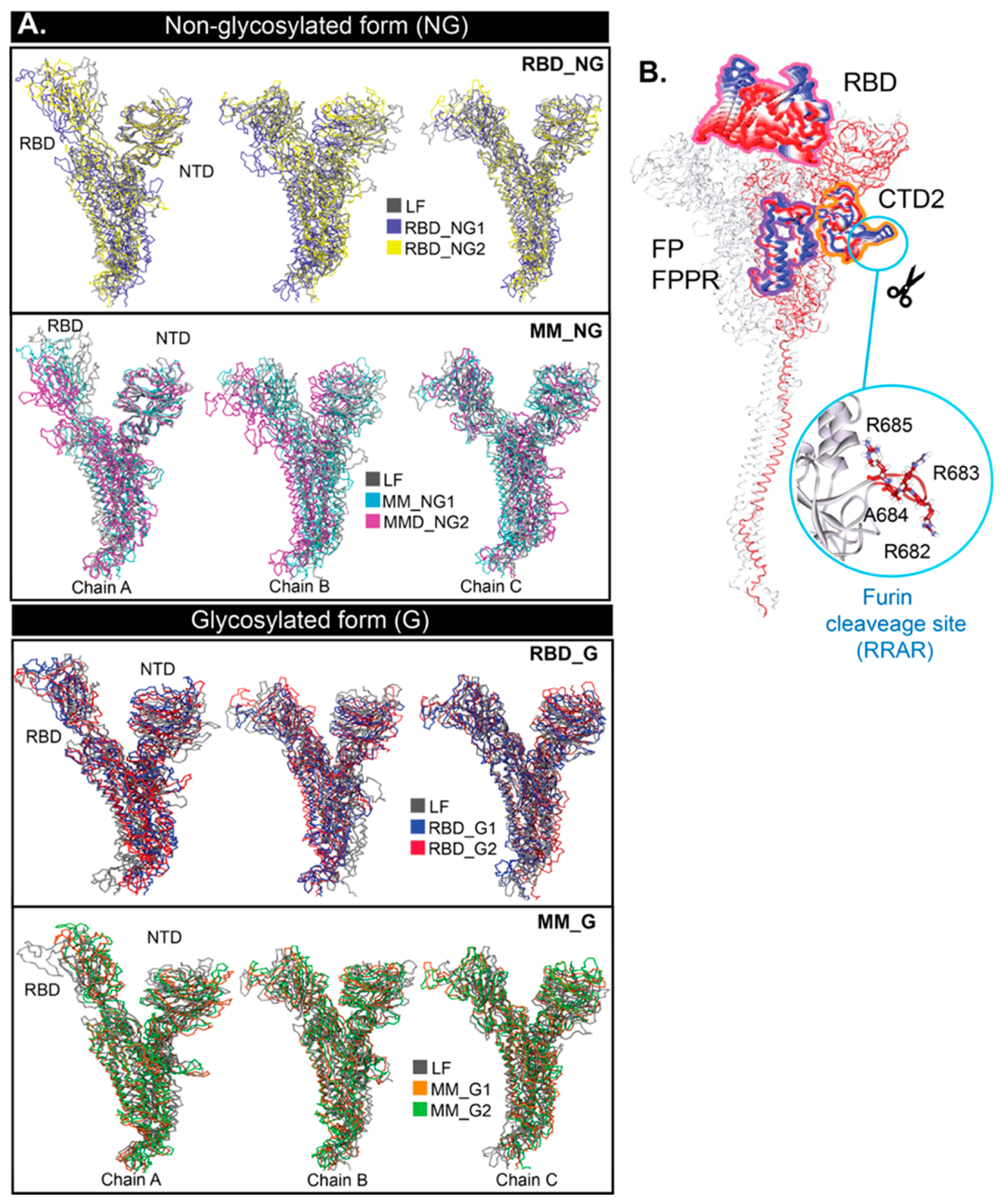
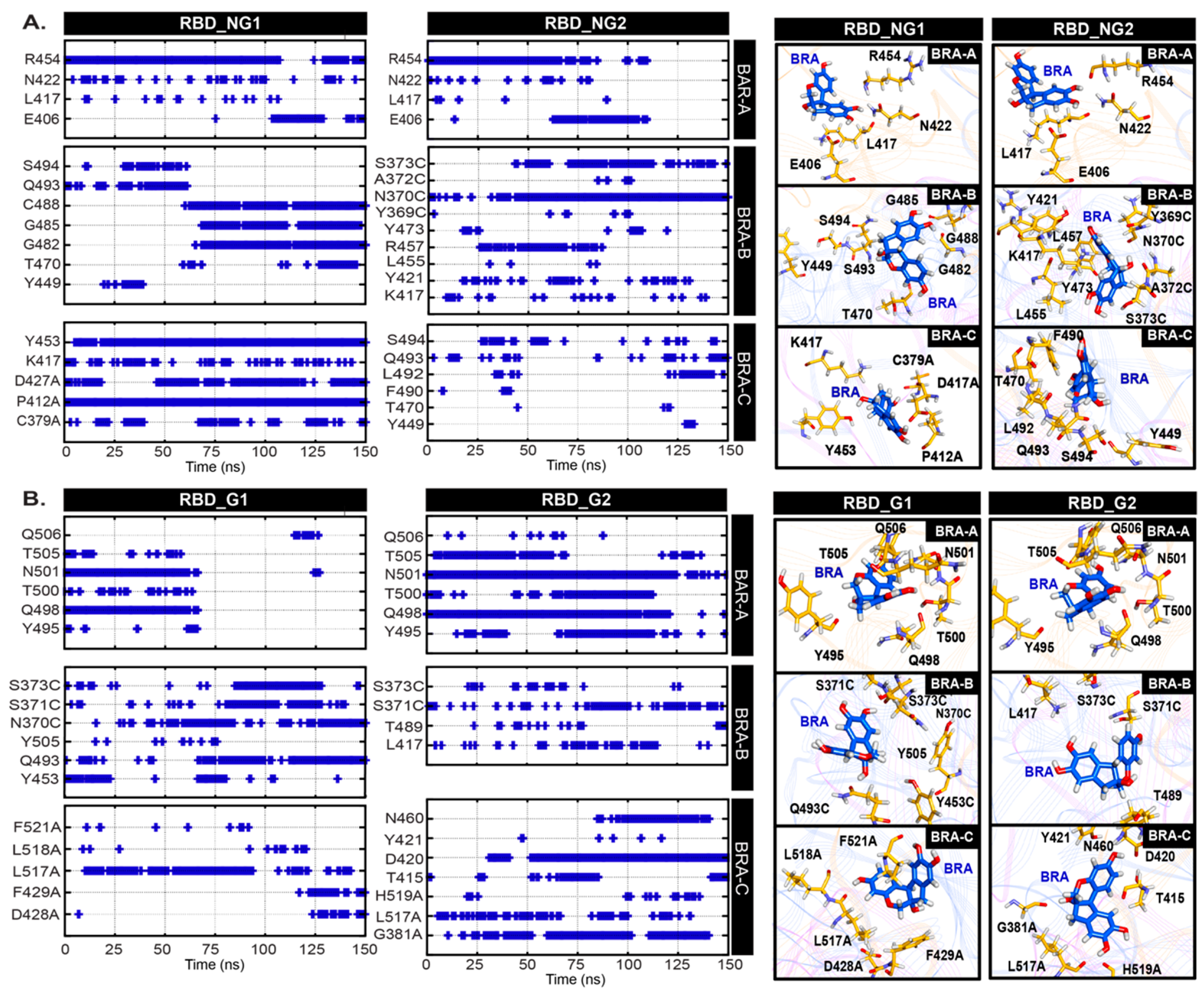
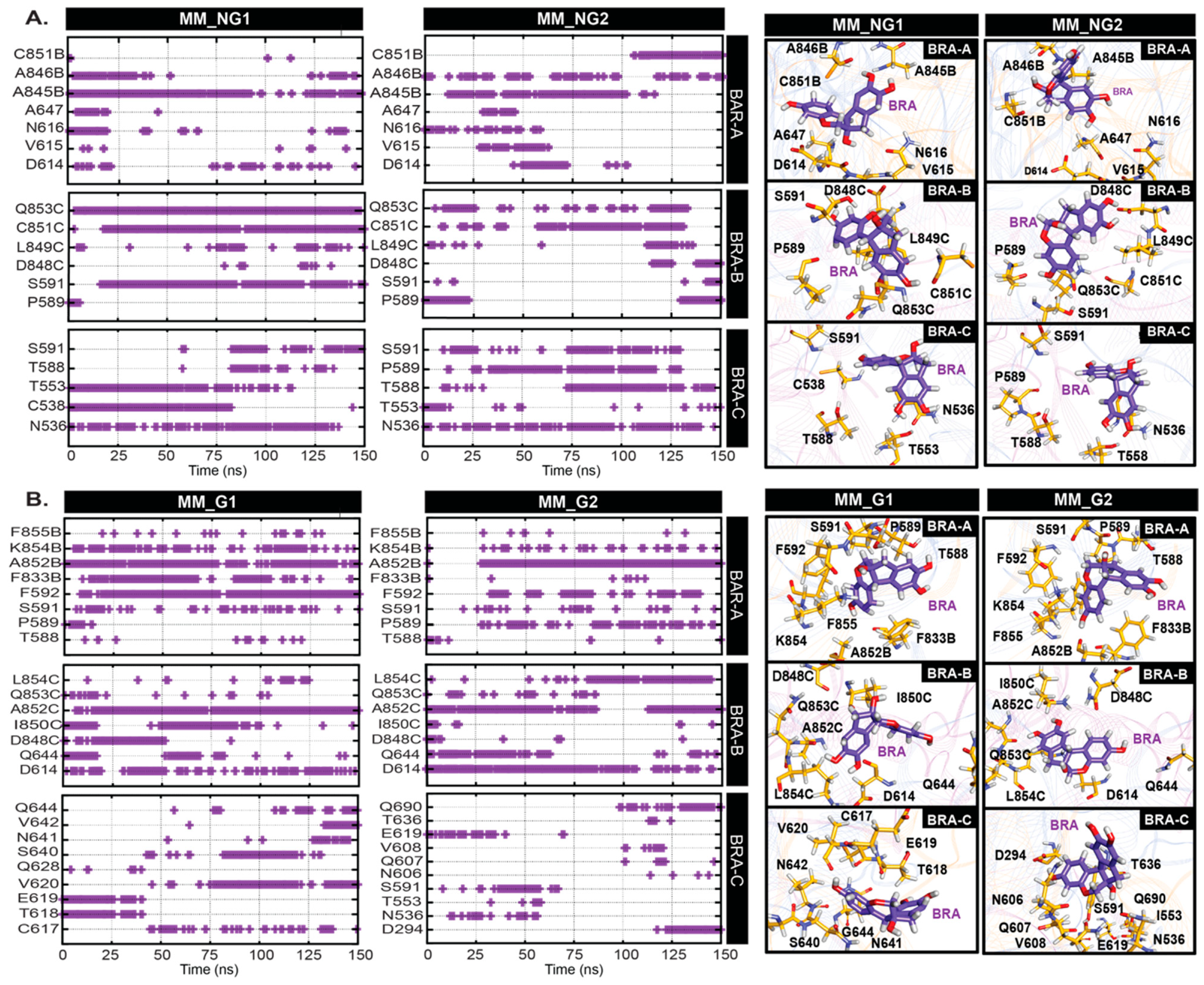
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MD | Molecular Dynamics |
| BRA | Brazilin |
References
- Huang, J.-T.; Ran, R.-X.; Lv, Z.-H.; Feng, L.-N.; Ran, C.-Y.; Tong, Y.-Q.; Li, D.; Su, H.-W.; Zhu, C.-L.; Qiu, S.-L. Chronological changes of viral shedding in adult inpatients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 2158–2166. [Google Scholar] [CrossRef] [PubMed]
- Kanjanasirirat, P.; Suksatu, A.; Manopwisedjaroen, S.; Munyoo, B.; Tuchinda, P.; Jearawuttanakul, K.; Seemakhan, S.; Charoensutthivarakul, S.; Wongtrakoongate, P.; Rangkasenee, N. High-content screening of Thai medicinal plants reveals Boesenbergia rotunda extract and its component Panduratin A as anti-SARS-CoV-2 agents. Sci. Rep. 2020, 10, 19963. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.; Baker, S.; Baric, R.S.; de Groot, R.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.; Lauber, C.; Leontovich, A.M.; Neuman, B.W. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Jan, J.-T.; Cheng, T.-J.R.; Juang, Y.-P.; Ma, H.-H.; Wu, Y.-T.; Yang, W.-B.; Cheng, C.-W.; Chen, X.; Chou, T.-H.; Shie, J.-J. Identification of existing pharmaceuticals and herbal medicines as inhibitors of SARS-CoV-2 infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2021579118. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Zuzic, L.; Samsudin, F.; Shivgan, A.T.; Raghuvamsi, P.V.; Marzinek, J.K.; Boags, A.; Pedebos, C.; Tulsian, N.K.; Warwicker, J.; MacAry, P.; et al. Uncovering cryptic pockets in the SARS-CoV-2 spike glycoprotein. Structure 2022, 30, 1062–1074.e4. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Zhao, X.; Chen, H.; Wang, H. Glycans of SARS-CoV-2 spike protein in virus infection and antibody production. Front. Mol. Biosci. 2021, 8, 629873. [Google Scholar] [CrossRef]
- Li, Y.; Liu, D.; Wang, Y.; Su, W.; Liu, G.; Dong, W. The importance of glycans of viral and host proteins in enveloped virus infection. Front. Immunol. 2021, 12, 638573. [Google Scholar] [CrossRef]
- Zhang, F.; Schmidt, F.; Muecksch, F.; Wang, Z.; Gazumyan, A.; Nussenzweig, M.C.; Gaebler, C.; Caskey, M.; Hatziioannou, T.; Bieniasz, P.D. SARS-CoV-2 spike glycosylation affects function and neutralization sensitivity. Mbio 2024, 15, e01672-23. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, G.R. SARS-CoV-2 spike and its adaptable furin cleavage site. Lancet Microbe 2021, 2, e488–e489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 2021, 372, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.L.; Quirante, J.C.; Vargas, L.E.Z.; Gatchalian, A.F.; Nellas, R.B. Complementary Pocket and Network-Based Approach to Search for Spike Protein Allosteric Pocket Sites. ACS Omega 2023, 8, 45313–45325. [Google Scholar] [CrossRef]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Kavanagh Williamson, M.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020, 370, 725–730. [Google Scholar] [CrossRef]
- Gupta, G.; Verkhivker, G. Exploring Binding Pockets in the Conformational States of the SARS-CoV-2 Spike Trimers for the Screening of Allosteric Inhibitors Using Molecular Simulations and Ensemble-Based Ligand Docking. Int. J. Mol. Sci. 2024, 25, 4955. [Google Scholar] [CrossRef]
- Bangaru, S.; Ozorowski, G.; Turner, H.L.; Antanasijevic, A.; Huang, D.; Wang, X.; Torres, J.L.; Diedrich, J.K.; Tian, J.-H.; Portnoff, A.D. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science 2020, 370, 1089–1094. [Google Scholar] [CrossRef]
- Carrique, L.; Duyvesteyn, H.M.; Malinauskas, T.; Zhao, Y.; Ren, J.; Zhou, D.; Walter, T.S.; Radecke, J.; Huo, J.; Ruza, R.R. The SARS-CoV-2 Spike harbours a lipid binding pocket which modulates stability of the prefusion trimer. BioRxiv 2020. [Google Scholar] [CrossRef]
- Rosa, A.; Pye, V.; Graham, C. SARS-CoV-2 recruits a haem metabolite to evade antibody immunity. medRxiv Prepr. Serv. Heal. Sci. 2021. preprint. [Google Scholar]
- Ma, B.; Zhang, Z.; Li, Y.; Lin, X.; Gu, N. Evaluation of interactions between SARS-CoV-2 RBD and full-length ACE2 with coarse-grained molecular dynamics simulations. J. Chem. Inf. Model. 2022, 62, 936–944. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Key interacting residues between RBD of SARS-CoV-2 and ACE2 receptor: Combination of molecular dynamics simulation and density functional calculation. J. Chem. Inf. Model. 2021, 61, 4425–4441. [Google Scholar] [CrossRef]
- Drożdżal, S.; Rosik, J.; Lechowicz, K.; Machaj, F.; Kotfis, K.; Ghavami, S.; Łos, M.J. FDA approved drugs with pharmacotherapeutic potential for SARS-CoV-2 (COVID-19) therapy. Drug Resist. Updates 2020, 53, 100719. [Google Scholar] [CrossRef]
- Feng, B.; Fu, K. Latest development of approved COVID-19 drugs and COVID-19 drugs undergoing late stage clinical trials. Front. Drug Discov. 2023, 3, 1304129. [Google Scholar] [CrossRef]
- Al-Kuraishy, H.M.; Al-Fakhrany, O.M.; Elekhnawy, E.; Al-Gareeb, A.I.; Alorabi, M.; De Waard, M.; Albogami, S.M.; Batiha, G.E.-S. Traditional herbs against COVID-19: Back to old weapons to combat the new pandemic. Eur. J. Med. Res. 2022, 27, 186. [Google Scholar] [CrossRef]
- Pham, T.X.; Huynh, T.T.; Kim, B.; Lim, Y.-S.; Hwang, S.B. A natural product YSK-A blocks SARS-CoV-2 propagation by targeting multiple host genes. Sci. Rep. 2023, 13, 21489. [Google Scholar] [CrossRef]
- Mukherjee, P.K.; Efferth, T.; Das, B.; Kar, A.; Ghosh, S.; Singha, S.; Debnath, P.; Sharma, N.; Bhardwaj, P.K.; Haldar, P.K. Role of medicinal plants in inhibiting SARS-CoV-2 and in the management of post-COVID-19 complications. Phytomedicine 2022, 98, 153930. [Google Scholar] [CrossRef]
- Isidoro, C.; Chang, A.C.-F.; Sheen, L.-Y. Natural products as a source of novel drugs for treating SARS-CoV2 infection. J. Tradit. Complement. Med. 2022, 12, 1–5. [Google Scholar] [CrossRef]
- Zahra, W.; Rai, S.N.; Birla, H.; Singh, S.S.; Rathore, A.S.; Dilnashin, H.; Keswani, C.; Singh, S.P. Economic importance of medicinal plants in Asian countries. In Bioeconomy for Sustainable Development; Springer: Singapore, 2020; pp. 359–377. [Google Scholar]
- Asigbaase, M.; Adusu, D.; Anaba, L.; Abugre, S.; Kang-Milung, S.; Acheamfour, S.A.; Adamu, I.; Ackah, D.K. Conservation and economic benefits of medicinal plants: Insights from forest-fringe communities of Southwestern Ghana. Trees For. People 2023, 14, 100462. [Google Scholar] [CrossRef]
- Moyeenudin, H.; Thiruchelvi, R.; Lawrence, A.W.; Williams, R.J. The phytochemical components of caesalpinia sappan in treating respiratory ailments through an herbal soup in addition with sensory evaluation. Mater. Today Proc. 2022, 56, 2167–2171. [Google Scholar] [CrossRef]
- Arjin, C.; Tateing, S.; Potapohn, N.; Arunorat, J.; Pringproa, K.; Lumsangkul, C.; Seel-Audom, M.; Ruksiriwanich, W.; Sringarm, K. Brazilin from Caesalpinia sappan inhibits viral infection against PRRSV via CD163ΔSRCR5 MARC-145 cells: An in silico and in vitro studies. Sci. Rep. 2022, 12, 21595. [Google Scholar] [CrossRef]
- Arjin, C.; Hongsibsong, S.; Pringproa, K.; Seel-Audom, M.; Ruksiriwanich, W.; Sutan, K.; Sommano, S.R.; Sringarm, K. Effect of Ethanolic Caesalpinia sappan Fraction on In Vitro Antiviral Activity against Porcine Reproductive and Respiratory Syndrome Virus. Vet. Sci. 2021, 8, 106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. [Google Scholar] [CrossRef]
- Pal, D. Spike protein fusion loop controls SARS-CoV-2 fusogenicity and infectivity. J. Struct. Biol. 2021, 213, 107713. [Google Scholar] [CrossRef]
- Lemmin, T.; Kalbermatter, D.; Harder, D.; Plattet, P.; Fotiadis, D. Structures and dynamics of the novel S1/S2 protease cleavage site loop of the SARS-CoV-2 spike glycoprotein. J. Struct. Biol. X 2020, 4, 100038. [Google Scholar] [CrossRef]
- Wang, S.; Ran, W.; Sun, L.; Fan, Q.; Zhao, Y.; Wang, B.; Yang, J.; He, Y.; Wu, Y.; Wang, Y. Sequential glycosylations at the multibasic cleavage site of SARS-CoV-2 spike protein regulate viral activity. Nat. Commun. 2024, 15, 4162. [Google Scholar] [CrossRef]
- Zhang, L.; Mann, M.; Syed, Z.A.; Reynolds, H.M.; Tian, E.; Samara, N.L.; Zeldin, D.C.; Tabak, L.A.; Ten Hagen, K.G. Furin cleavage of the SARS-CoV-2 spike is modulated by O-glycosylation. Proc. Natl. Acad. Sci. USA 2021, 118, e2109905118. [Google Scholar] [CrossRef]
- Beaudoin, C.A.; Pandurangan, A.P.; Kim, S.Y.; Hamaia, S.W.; Huang, C.L.H.; Blundell, T.L.; Vedithi, S.C.; Jackson, A.P. In silico analysis of mutations near S1/S2 cleavage site in SARS-CoV-2 spike protein reveals increased propensity of glycosylation in Omicron strain. J. Med. Virol. 2022, 94, 4181–4192. [Google Scholar] [CrossRef]
- Du, Y.; Wang, H.; Chen, L.; Fang, Q.; Zhang, B.; Jiang, L.; Wu, Z.; Yang, Y.; Zhou, Y.; Chen, B. Non-RBM mutations impaired SARS-CoV-2 spike protein regulated to the ACE2 receptor based on molecular dynamic simulation. Front. Mol. Biosci. 2021, 8, 614443. [Google Scholar] [CrossRef]
- Qu, K.; Xiong, X.; Ciazynska, K.A.; Carter, A.P.; Briggs, J.A. Structures and function of locked conformations of SARS-CoV-2 spike. BioRxiv 2021. preprint. [Google Scholar]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Rangarajan, E.S.; Izard, T.; Farzan, M.; Choe, H. The D614G mutation in the SARS-CoV-2 spike protein reduces S1 shedding and increases infectivity. BioRxiv 2020. preprint. [Google Scholar]
- Jackson, C.B.; Zhang, L.; Farzan, M.; Choe, H. Functional importance of the D614G mutation in the SARS-CoV-2 spike protein. Biochem. Biophys. Res. Commun. 2021, 538, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B. PubChem 2023 update. Nucleic Acids Res. 2023, 51, D1373–D1380. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking11Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. Mol. Model. Annu. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Jo, S.; MacKerell, A.D., Jr.; Klauda, J.B.; Im, W. CHARMM-GUI Input Generator for NAMD, Gromacs, Amber, Openmm, and CHARMM/OpenMM Simulations using the CHARMM36 Additive Force Field. Biophys. J. 2016, 110, 641a. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R., 3rd; McGee, T.D., Jr.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Number of Hydrogen Bonds | |||||
|---|---|---|---|---|---|---|
| Water | Protein | |||||
| BRA-A | BRA-B | BRA-C | BRA-A | BRA-B | BRA-C | |
| MM_NG1 | 5.6 ± 1.5 | 4.6 ± 1.4 | 5.5 ± 1.6 | 1.5 ± 1.1 | 2.48 ± 0.73 | 1.3 ± 0.9 |
| MM_NG2 | 5.4 ± 1.5 | 4.9 ± 1.6 | 4.9 ± 1.5 | 1.3 ± 0.9 | 1.53 ± 1.18 | 1.9 ± 1.1 |
| MM_G1 | 4.0 ± 1.3 | 3.7 ± 1.4 | 4.0 ± 1.7 | 1.5 ± 0.8 | 1.66 ± 0.87 | 1.3 ± 0.9 |
| MM_G2 | 4.4 ± 1.4 | 4.1 ± 1.6 | 5.6 ± 1.5 | 1.2 ± 0.8 | 2.14 ± 1.15 | 0.8 ± 0.7 |
| RBD_NG1 | 5.2 ± 1.3 | 5.1 ± 1.7 | 3.8 ± 1.3 | 0.7 ± 0.6 | 1.46 ± 1.07 | 2.4 ± 0.8 |
| RBD_NG2 | 5.0 ± 1.4 | 4.8 ± 1.6 | 5.8 ± 1.5 | 0.8 ± 0.6 | 1.04 ± 0.76 | 0.5 ± 0.7 |
| RBD_G1 | 5.7 ± 1.5 | 4.4 ± 1.7 | 5.8 ± 1.5 | 1.5 ± 1.0 | 1.18 ± 0.89 | 0.6 ± 0.6 |
| RBD_G2 | 4.4 ± 1.6 | 4.7 ± 1.5 | 4.4 ± 1.6 | 2.4 ± 1.4 | 0.39 ± 0.59 | 1.8 ± 0.9 |
| System | BRA-A | BRA-B | BRA-C | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ΔEvdw | ΔEElec | Total Energy | ΔEvdw | ΔEElec | Total Energy | ΔEvdw | ΔEElec | Total Energy | |
| MM_NG1 | −127.6 | −45.3 | −172.9 | −125.0 | −120.4 | −245.4 | −63.4 | −83.0 | −146.3 |
| MM_NG2 | −106.3 | −45.4 | −151.7 | −76.5 | −96.5 | −173.0 | 88.0 | −67.0 | −155.0 |
| MM_G1 | −126.7 | −93.1 | −219.8 | −102.8 | −46.8 | −149.6 | −77.4 | −49.6 | −127.0 |
| MM_G2 | −102.7 | −83.0 | −185.7 | −122.3 | −132.0 | −254.3 | −83.3 | −59.6 | −143.0 |
| RBD_NG1 | −88.8 | −70.3 | −159.1 | −85.6 | −71.3 | −156.9 | −109.3 | −139.5 | −248.8 |
| RBD_NG2 | −81.6 | −44.5 | −126.2 | −122.0 | −38.9 | −160.9 | −71.4 | −15.7 | −87.1 |
| RBD_G1 | −51.8 | −29.3 | −81.1 | −59.1 | −53.5 | −112.6 | −77.4 | −37.0 | −114.4 |
| RBD_G2 | −83.5 | −50.8 | −134.4 | −90.2 | −20.0 | −110.2 | −106.4 | −116.5 | −222.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bamrung, P.; Toviwek, B.; Samsudin, F.; Chairatana, P.; Bond, P.J.; Pongprayoon, P. The Binding of Brazilin from C. sappan to the Full-Length SARS-CoV-2 Spike Proteins. Int. J. Mol. Sci. 2025, 26, 4100. https://doi.org/10.3390/ijms26094100
Bamrung P, Toviwek B, Samsudin F, Chairatana P, Bond PJ, Pongprayoon P. The Binding of Brazilin from C. sappan to the Full-Length SARS-CoV-2 Spike Proteins. International Journal of Molecular Sciences. 2025; 26(9):4100. https://doi.org/10.3390/ijms26094100
Chicago/Turabian StyleBamrung, Phonphiphat, Borvornwat Toviwek, Firdaus Samsudin, Phoom Chairatana, Peter John Bond, and Prapasiri Pongprayoon. 2025. "The Binding of Brazilin from C. sappan to the Full-Length SARS-CoV-2 Spike Proteins" International Journal of Molecular Sciences 26, no. 9: 4100. https://doi.org/10.3390/ijms26094100
APA StyleBamrung, P., Toviwek, B., Samsudin, F., Chairatana, P., Bond, P. J., & Pongprayoon, P. (2025). The Binding of Brazilin from C. sappan to the Full-Length SARS-CoV-2 Spike Proteins. International Journal of Molecular Sciences, 26(9), 4100. https://doi.org/10.3390/ijms26094100