
In Silico Insights into the Inhibition of ADAMTS-5 by Punicalagin and Ellagic Acid for the Treatment of Osteoarthritis

Abstract
1. Introduction
2. Results
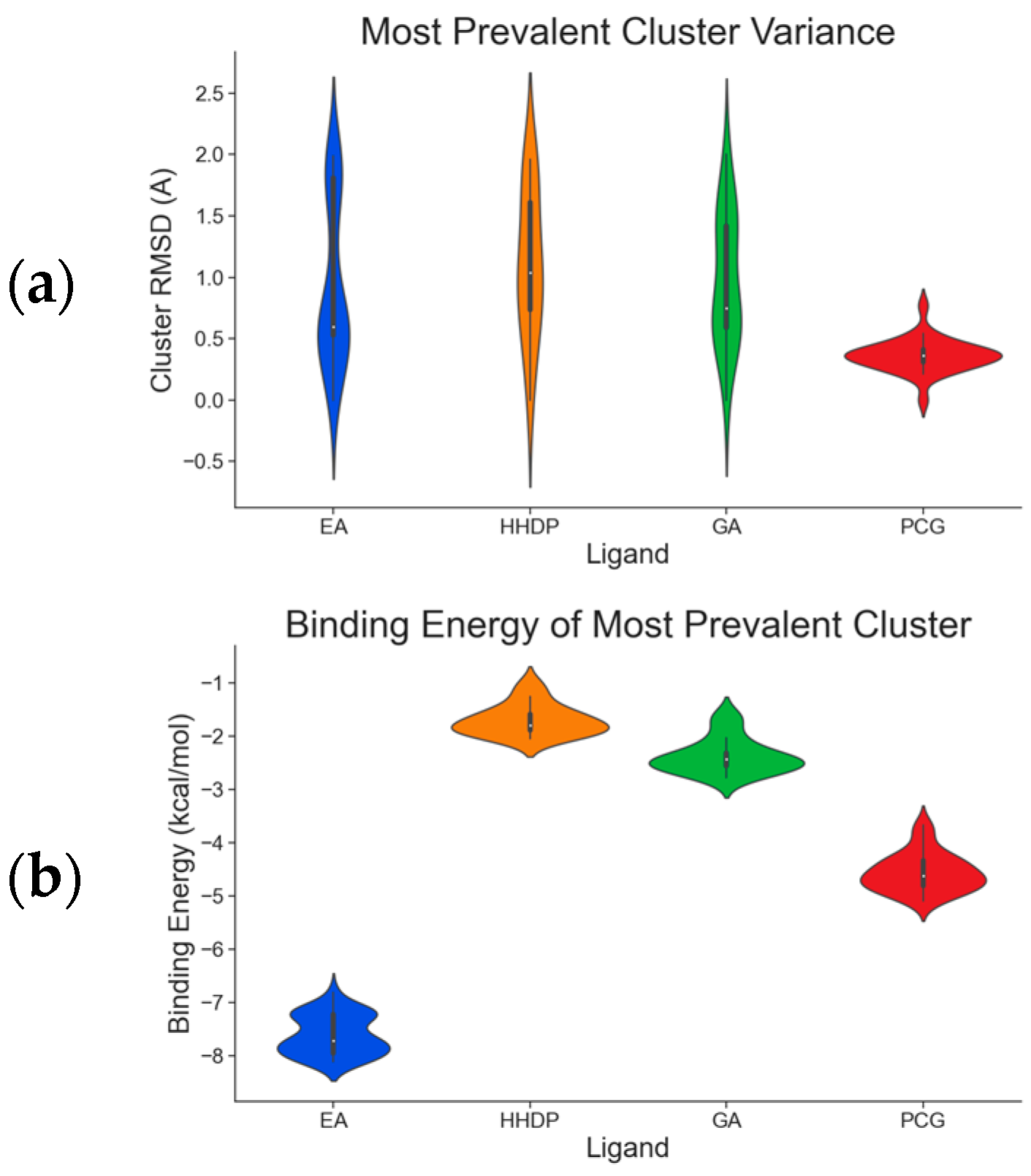
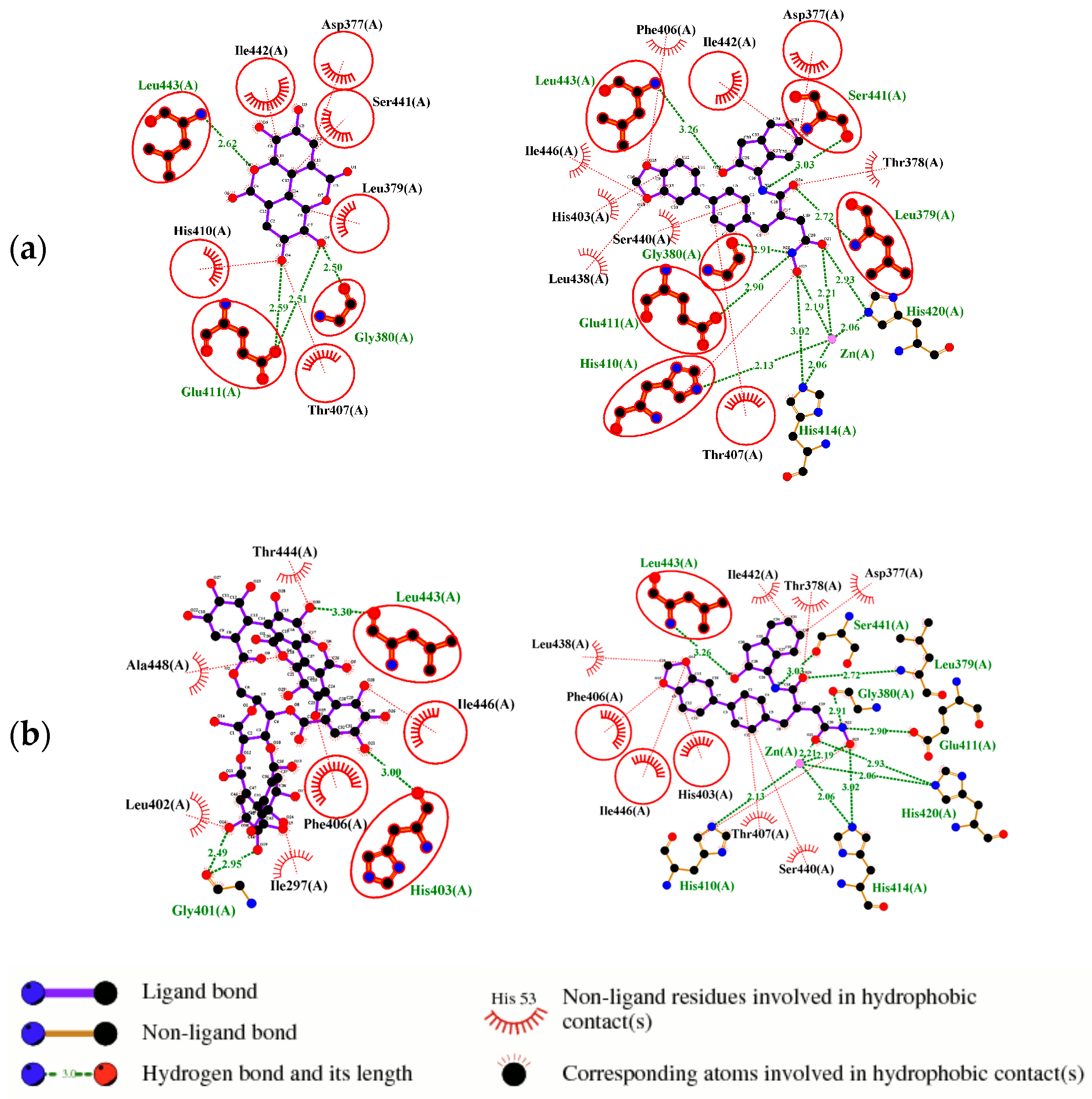
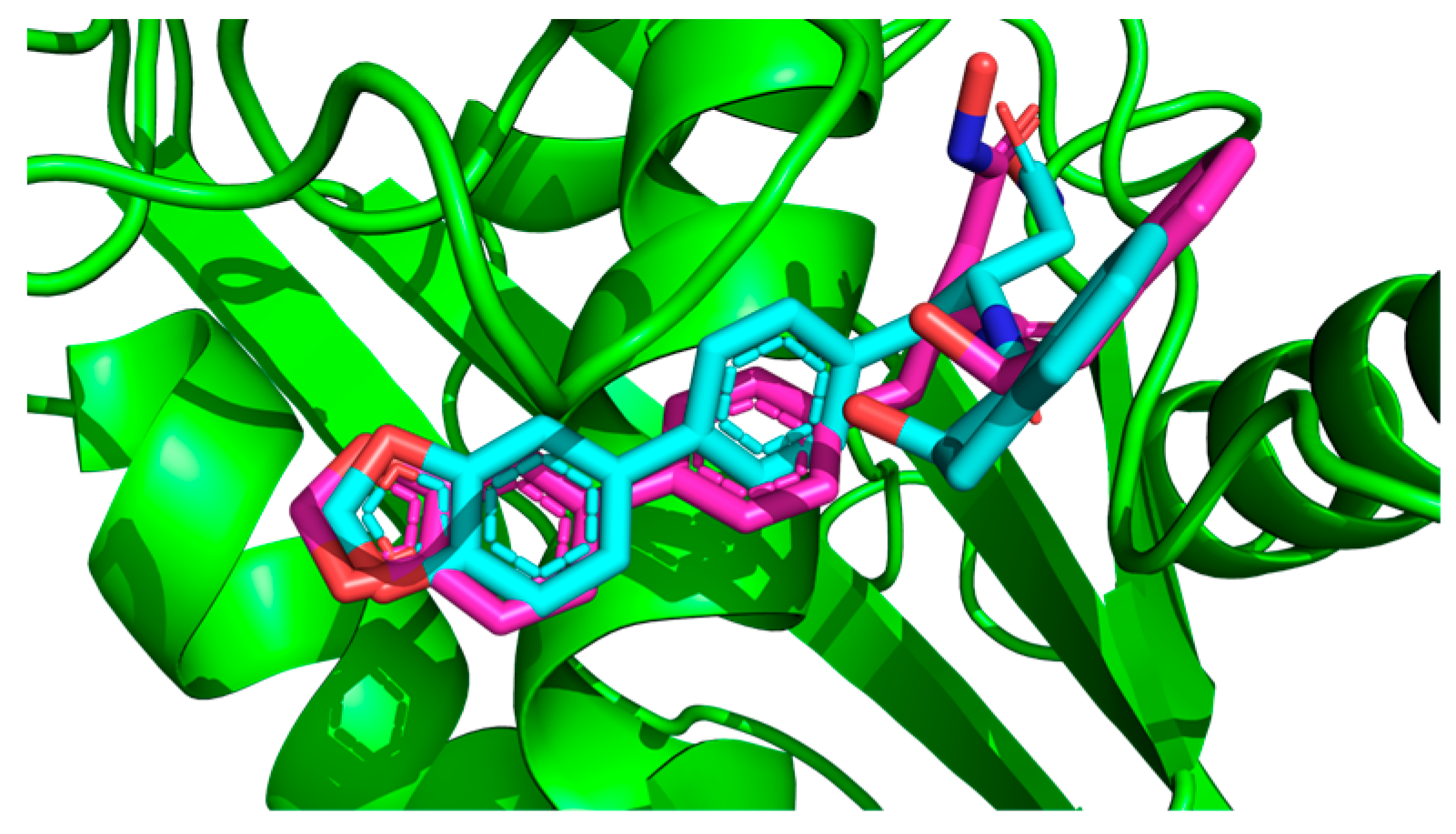
2.1. Molecular Docking Simulations
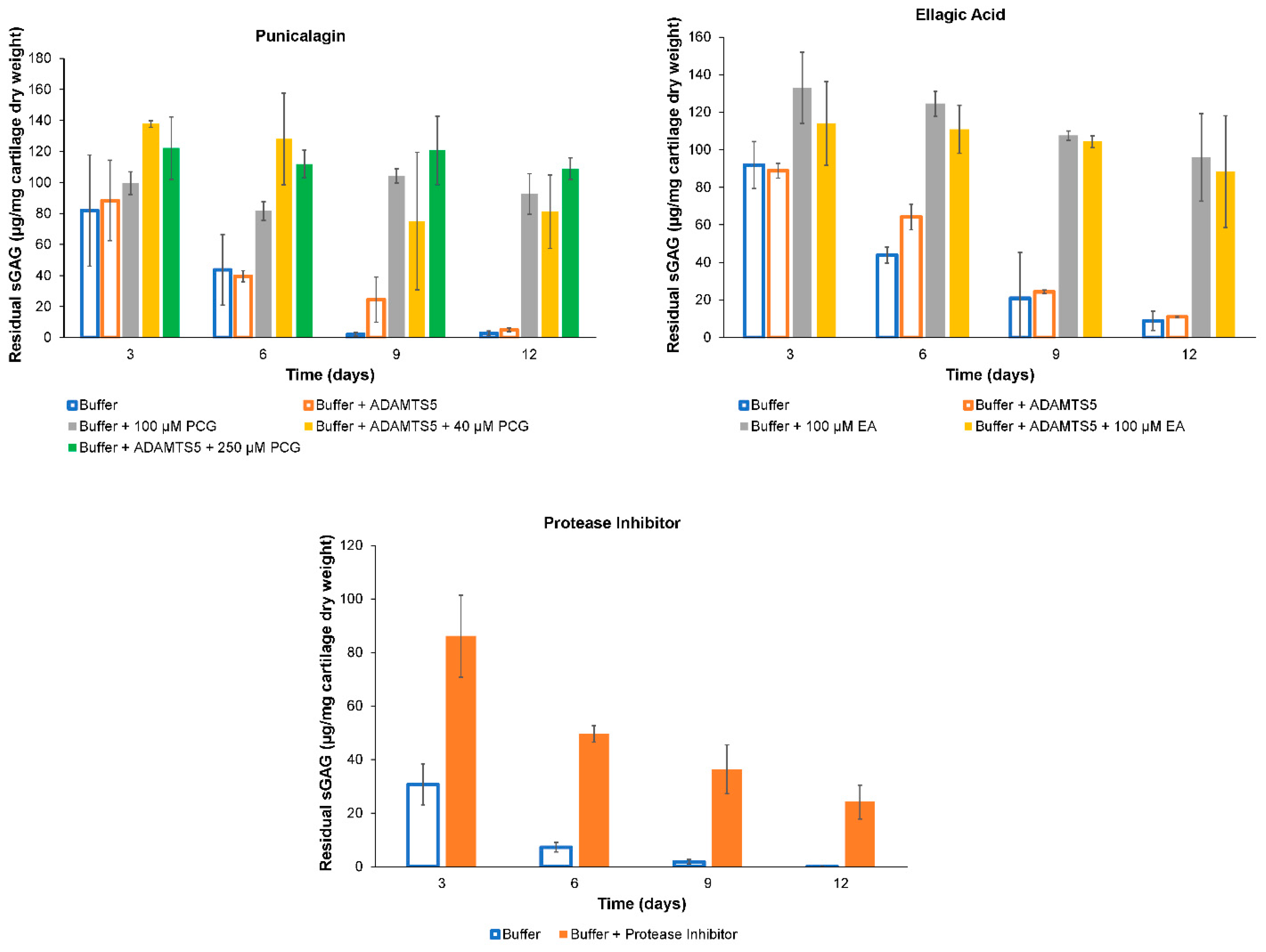
2.2. ADAMTS-5 Inhibition
2.3. Histology
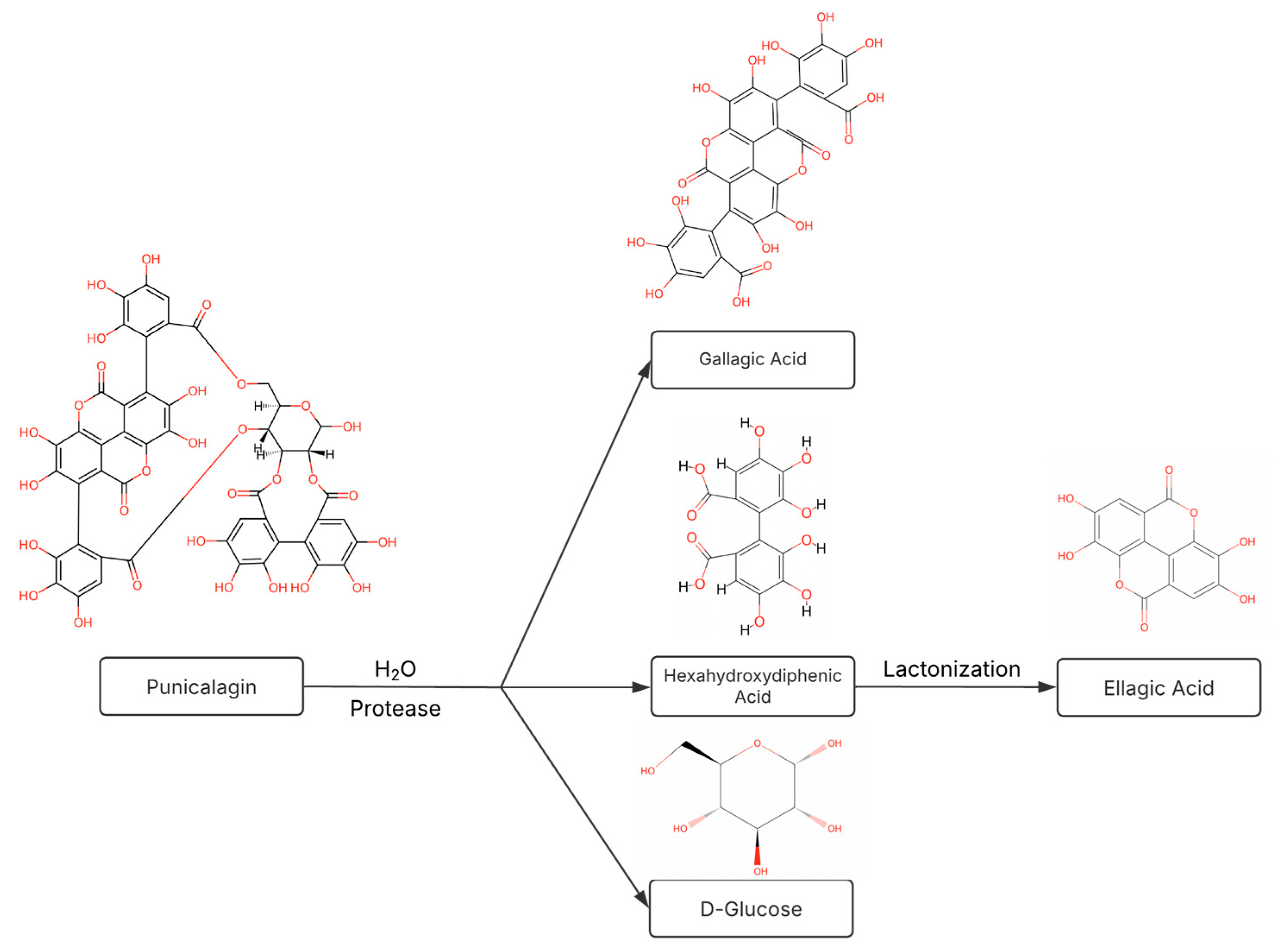
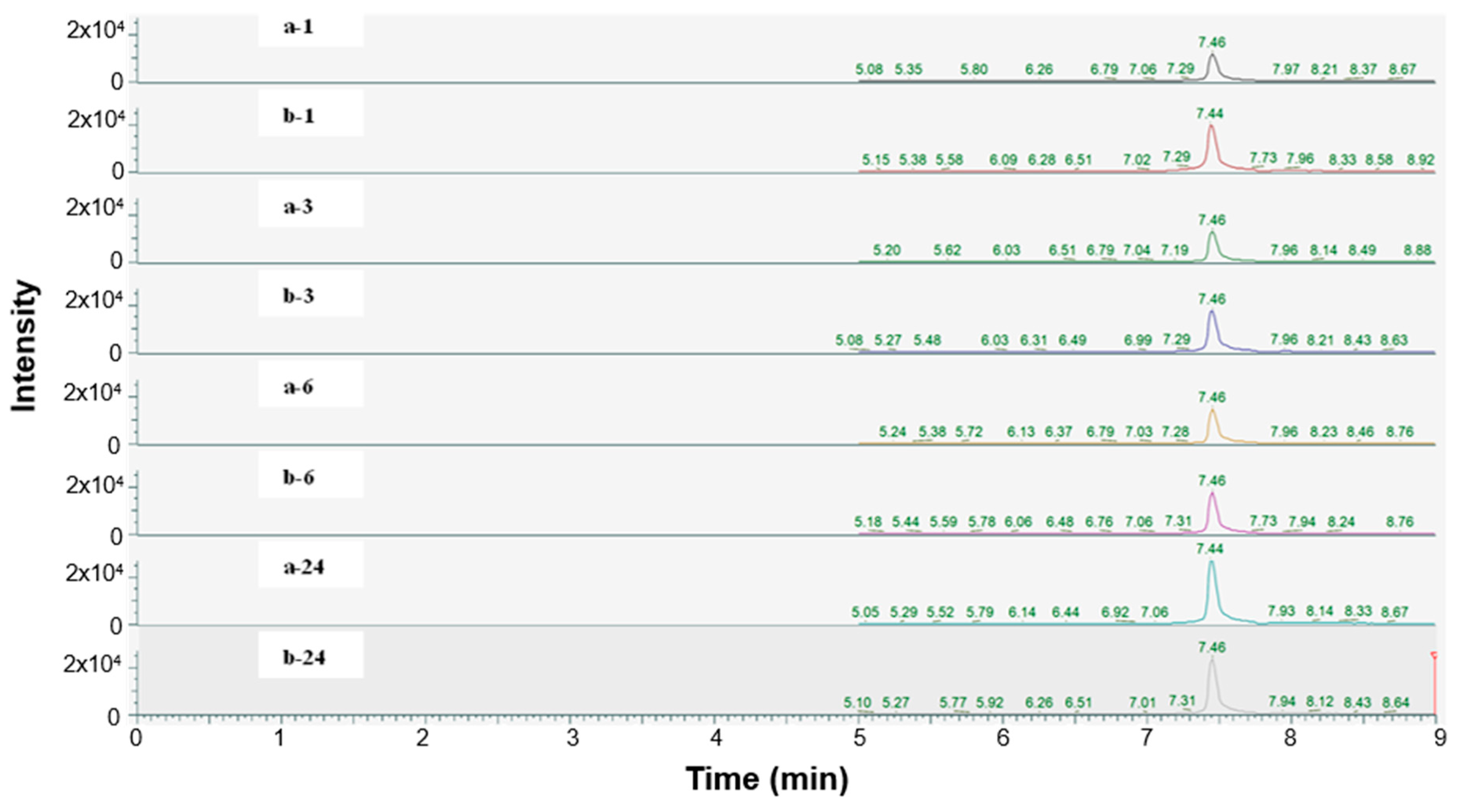
2.4. Hydrolyzability of Punicalagin
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Ligand Characterization
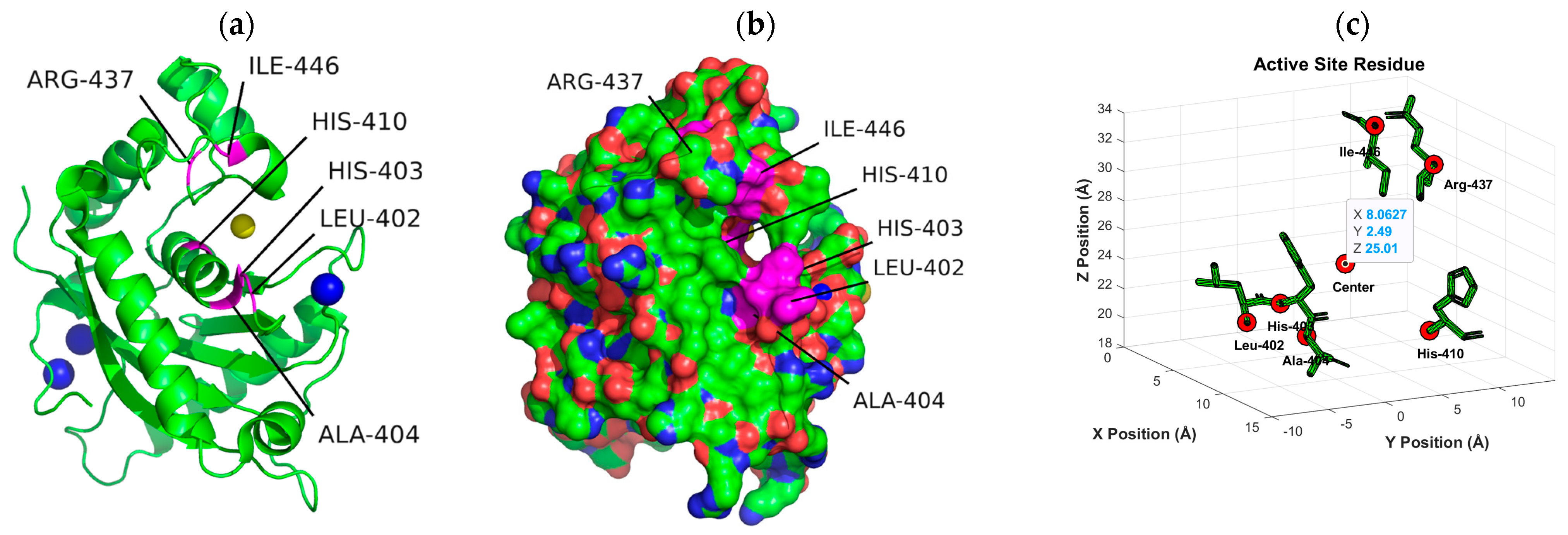
4.3. Enzyme Pocket Characterization
4.4. Molecular Docking Simulations
4.5. ADAMTS-5 Inhibition
4.6. Histology
4.7. Hydrolyzability of Punicalagin
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| OA | Osteoarthritis |
| PCG | Punicalagin |
| ECM | Extracellular matrix |
| EA | Ellagic acid |
| DMOAD | Disease modifying osteoarthritis drug |
| sGAG | Sulfated glycosaminoglycan |
| IA | Intra-articular |
| MAPK | Mitogen-activated protein kinase |
| ADAMTS | A disintegrin-like and metalloprotease domain with thrombospondin type 1 repeats |
| HHDP | Hexahydroxydiphenic |
| GA | Gallagic acid |
| PSA | Polar surface area |
| A2N | GLPG1972/S201086 |
| ZBG | Zinc-binding group |
| DMSO | Dimethyl sulfoxide |
| RMSD | Root means square difference |
| PDB | Protein Data Bank |
| LC-MS | Liquid chromatography–mass spectrometry |
| SRM | Selected reaction monitoring |
References
- Long, H.; Liu, Q.; Yin, H.; Wang, K.; Diao, N.; Zhang, Y.; Lin, J.; Guo, A. Prevalence Trends of Site-Specific Osteoarthritis from 1990 to 2019: Findings from the Global Burden of Disease Study 2019. Arthritis Rheumatol. 2022, 74, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- DeRogatis, M.; Anis, H.K.; Sodhi, N.; Ehiorobo, J.O.; Chughtai, M.; Bhave, A.; Mont, M.A. Non-operative treatment options for knee osteoarthritis. Ann. Transl. Med. 2019, 7 (Suppl. 7), S245. [Google Scholar] [CrossRef]
- Courties, A.; Kouki, I.; Soliman, N.; Mathieu, S.; Sellam, J. Osteoarthritis year in review 2024: Epidemiology and therapy. Osteoarthr. Cartil. 2024, 32, 1397–1404. [Google Scholar] [CrossRef]
- Aubourg, G.; Rice, S.J.; Bruce-Wootton, P.; Loughlin, J. Genetics of osteoarthritis. Osteoarthr. Cartil. 2022, 30, 636–649. [Google Scholar] [CrossRef] [PubMed]
- Nedunchezhiyan, U.; Varughese, I.; Sun, A.R.J.; Wu, X.; Crawford, R.; Prasadam, I. Obesity, Inflammation, and Immune System in Osteoarthritis. Front. Immunol. 2022, 13, 907750. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.M.; Cicuttini, F.M.; Alyousef, B.; Wang, Y. Female hormonal factors and osteoarthritis of the knee, hip and hand: A narrative review. Climacteric 2018, 21, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Neuprez, A.; Neuprez, A.H.; Kaux, J.F.; Kurth, W.; Daniel, C.; Thirion, T.; Huskin, J.P.; Gillet, P.; Bruyère, O.; Reginster, J.Y. Total joint replacement improves pain, functional quality of life, and health utilities in patients with late-stage knee and hip osteoarthritis for up to 5 years. Clin. Rheumatol. 2020, 39, 861–871. [Google Scholar] [CrossRef]
- Tuthill, T.; Jackson, G.R.; Schundler, S.F.; Lee, J.S.; Allahabadi, S.; Salazar, L.M.; McCormick, J.R.; Jawanda, H.; Batra, A.; Khan, Z.A.; et al. Radiofrequency Chondroplasty of the Knee Yields Excellent Clinical Outcomes and Minimal Complications: A Systematic Review. Arthrosc. Sports Med. Rehabil. 2023, 5, 100749. [Google Scholar] [CrossRef]
- Trofa, D.P.; Hong, I.S.; Lopez, C.D.; Rao, A.J.; Yu, Z.; Odum, S.M.; Moorman, C.T.; Piasecki, D.P.; Fleischli, J.E.; Saltzman, B.M. Isolated Osteochondral Autograft Versus Allograft Transplantation for the Treatment of Symptomatic Cartilage Lesions of the Knee: A Systematic Review and Meta-analysis. Am. J. Sports Med. 2023, 51, 812–824. [Google Scholar] [CrossRef]
- Foster, N.E.; Eriksson, L.; Deveza, L.; Hall, M. Osteoarthritis year in review 2022: Epidemiology & therapy. Osteoarthr. Cartil. 2023, 31, 876–883. [Google Scholar] [CrossRef]
- Katz, J.N.; Arant, K.R.; Loeser, R.F. Diagnosis and Treatment of Hip and Knee Osteoarthritis: A Review. Jama 2021, 325, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Elder, S.H.; Mosher, M.L.; Jarquin, P.; Smith, P.; Chironis, A. Effects of short-duration treatment of cartilage with punicalagin and genipin and the implications for treatment of osteoarthritis. J. Biomed. Mater. Res. Part B Appl. Biomater. 2021, 109, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Elder, S.H.; Ross, M.K.; Nicaise, A.J.; Miller, I.N.; Breland, A.N.; Hood, A.R.S. Development of in situ forming implants for controlled delivery of punicalagin. Int. J. Pharm. 2024, 652, 123842. [Google Scholar] [CrossRef]
- Xu, J.; Cao, K.; Liu, X.; Zhao, L.; Feng, Z.; Liu, J. Punicalagin Regulates Signaling Pathways in Inflammation-Associated Chronic Diseases. Antioxidants 2021, 11, 29. [Google Scholar] [CrossRef]
- Cao, Y.; Chen, J.; Ren, G.; Zhang, Y.; Tan, X.; Yang, L. Punicalagin Prevents Inflammation in LPS- Induced RAW264.7 Macrophages by Inhibiting FoxO3a/Autophagy Signaling Pathway. Nutrients 2019, 11, 2794. [Google Scholar] [CrossRef]
- Lee, C.J.; Chen, L.G.; Liang, W.L.; Hsieh, M.S.; Wang, C.C. Inhibitory effects of punicalagin from Punica granatum against type II collagenase-induced osteoarthritis. J. Funct. Foods 2018, 41, 216–222. [Google Scholar] [CrossRef]
- Liu, F.; Yang, H.; Li, D.; Wu, X.; Han, Q. Punicalagin attenuates osteoarthritis progression via regulating Foxo1/Prg4/HIF3α axis. Bone 2021, 152, 116070. [Google Scholar] [CrossRef]
- Rozadi, N.; Oktavia, S.; Fauziah, F. Pharmacological Activities of Punicalagin: A Review. J. Drug Deliv. Ther. 2022, 12, 148–155. [Google Scholar] [CrossRef]
- Xu, X.; Yin, P.; Wan, C.; Chong, X.; Liu, M.; Cheng, P.; Chen, J.; Liu, F.; Xu, J. Punicalagin inhibits inflammation in LPS-induced RAW264.7 macrophages via the suppression of TLR4-mediated MAPKs and NF-κB activation. Inflammation 2014, 37, 956–965. [Google Scholar] [CrossRef]
- Attur, M.; Al-Mussawir, H.E.; Patel, J.; Kitay, A.; Dave, M.; Palmer, G.; Pillinger, M.H.; Abramson, S.B. Prostaglandin E2 Exerts Catabolic Effects in Osteoarthritis Cartilage: Evidence for Signaling via the EP4 Receptor. J. Immunol. 2008, 181, 5082–5088. [Google Scholar] [CrossRef]
- Jean-Gilles, D.; Li, L.; Vaidyanathan, V.; King, R.; Cho, B.; Worthen, D.R.; Chichester, C.O.; Seeram, N.P. Inhibitory effects of polyphenol punicalagin on type-II collagen degradation in vitro and inflammation in vivo. Chem. Interact. 2013, 205, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Khan, N.M.; Ashruf, O.S.; Haqqi, T.M. Inhibition of cartilage degradation and suppression of PGE2 and MMPs expression by pomegranate fruit extract in a model of posttraumatic osteoarthritis. Nutrition 2017, 33, 1–13. [Google Scholar] [CrossRef]
- Huang, M.; Wu, K.; Zeng, S.; Liu, W.; Cui, T.; Chen, Z.; Lin, L.; Chen, D.; Ouyang, H. Punicalagin inhibited inflammation and migration of fibroblast-like synoviocytes through nf-κb pathway in the experimental study of rheumatoid arthritis. J. Inflamm. Res. 2021, 14, 1901–1913. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.P.; Mahal, H.S.; Kapoor, S.; Aradhya, S.M. In vitro studies on the binding, antioxidant, and cytotoxic action of punicalagin. J. Agric. Food Chem. 2007, 55, 1491–1500. [Google Scholar] [CrossRef]
- Cerdá, B.; Cerón, J.J.; Tomás-Barberán, F.A.; Espín, J.C. Repeated oral administration of high doses of the pomegranate ellagitannin punicalagin to rats for 37 days is not toxic. J. Agric. Food Chem. 2003, 51, 3493–3501. [Google Scholar] [CrossRef] [PubMed]
- Troeberg, L.; Nagase, H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2012, 1824, 133–145. [Google Scholar] [CrossRef]
- Li, T.; Peng, J.; Li, Q.; Shu, Y.; Zhu, P.; Hao, L. The Mechanism and Role of ADAMTS Protein Family in Osteoarthritis. Biomolecules 2022, 12, 959. [Google Scholar] [CrossRef] [PubMed]
- Kiani, C.; Chen, L.; Wu, Y.J.; Yee, A.J.; Yang, B.B. Structure and function of aggrecan. Cell Res. 2002, 12, 19–32. [Google Scholar] [CrossRef]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.; Blanchet, T.; Ma, H.-L.; Flannery, C.R.; Peluso, D.; Kanki, K.; Yang, Z.; et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef]
- Hering, N.A.; Luettig, J.; Jebautzke, B.; Schulzke, J.D.; Rosenthal, R. The Punicalagin Metabolites Ellagic Acid and Urolithin A Exert Different Strengthening and Anti-Inflammatory Effects on Tight Junction-Mediated Intestinal Barrier Function In Vitro. Front. Pharmacol. 2021, 12, 610164. [Google Scholar] [CrossRef]
- Caballero, V.; Estévez, M.; Tomás-Barberán, F.A.; Morcuende, D.; Martín, I.; Delgado, J. Biodegradation of Punicalagin into Ellagic Acid by Selected Probiotic Bacteria: A Study of the Underlying Mechanisms by MS-Based Proteomics. J. Agric. Food Chem. 2022, 70, 16273–16285. [Google Scholar] [CrossRef] [PubMed]
- Sabzevari, A.G.; Sabahi, H. Montmorillonite an efficient oral nanocarrier for punicalagin-rich pomegranate peel extract: An in vitro study. J Drug Deliv Sci Technol, J. Drug Deliv. Sci. Technol. 2023, 86, 104713. [Google Scholar] [CrossRef]
- Aguilera-Carbo, A.; Augur, C.; Prado-Barragan, L.A.; Favela-Torres, E.; Aguilar, C.N. Microbial production of ellagic acid and biodegradation of ellagitannins. Appl. Microbiol. Biotechnol. 2008, 78, 189–199. [Google Scholar] [CrossRef]
- Klein, P.; Johe, P.; Wagner, A.; Jung, S.; Kühlborn, J.; Barthels, F.; Tenzer, S.; Distler, U.; Waigel, W.; Engels, B.; et al. New Cysteine Protease Inhibitors: Electrophilic (Het)arenes and Unexpected Prodrug Identification for the Trypanosoma Protease Rhodesain. Molecules 2020, 25, 1451. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Rauf, A.; Guo, Y.; Kang, X. Real-Time Label-Free Kinetics Monitoring of Trypsin-Catalyzed Ester Hydrolysis by a Nanopore Sensor. ACS Sens. 2019, 4, 2854–2857. [Google Scholar] [CrossRef]
- Aqil, F.; Vadhanam, M.V.; Gupta, R.C. Enhanced activity of punicalagin delivered via polymeric implants against benzo[a]pyrene-induced DNA adducts. Mutat. Res. Toxicol. Environ. Mutagen. 2012, 743, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Ríos, J.L.; Giner, R.M.; Marín, M.; Recio, M.C. A Pharmacological Update of Ellagic Acid. Planta Med. 2018, 84, 1068–1093. [Google Scholar] [CrossRef]
- Lin, Z.; Lin, C.; Fu, C.; Lu, H.; Jin, H.; Chen, Q.; Pan, J. The protective effect of Ellagic acid (EA) in osteoarthritis: An in vitro and in vivo study. Biomed. Pharmacother. 2020, 125, 109845. [Google Scholar] [CrossRef]
- Brebion, F.; Gosmini, R.; Deprez, P.; Varin, M.; Peixoto, C.; Alvey, L.; Jary, H.; Bienvenu, N.; Triballeau, N.; Blanque, R.; et al. Discovery of GLPG1972/S201086, a Potent, Selective, and Orally Bioavailable ADAMTS-5 Inhibitor for the Treatment of Osteoarthritis. J. Med. Chem. 2021, 64, 2937–2952. [Google Scholar] [CrossRef]
- Chow, Y.Y.; Chin, K.Y. The Role of Inflammation in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2020, 2020, 1–19. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 2.6 Schrödinger, LLC. Available online: https://pymol.org/ (accessed on 27 May 2024).
- Woodell-May, J.E.; Sommerfeld, S.D. Role of Inflammation and the Immune System in the Progression of Osteoarthritis. J. Orthop. Res. 2020, 38, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Cuffaro, D.; Ciccone, L.; Rossello, A.; Nuti, E.; Santamaria, S. Targeting Aggrecanases for Osteoarthritis Therapy: From Zinc Chelation to Exosite Inhibition. J. Med. Chem. 2022, 65, 13505–13532. [Google Scholar] [CrossRef]
- Young, R.J.; Green, D.V.S.; Luscombe, C.N.; Hill, A.P. Getting physical in drug discovery II: The impact of chromatographic hydrophobicity measurements and aromaticity. Drug Discov. Today 2011, 16, 822–830. [Google Scholar] [CrossRef]
- Nuti, E.; Cuffaro, D.; Bernardini, E.; Camodeca, C.; Panelli, L.; Chaves, S.; Ciccone, L.; Tepshi, L.; Vera, L.; Orlandini, E.; et al. Development of Thioaryl-Based Matrix Metalloproteinase-12 Inhibitors with Alternative Zinc-Binding Groups: Synthesis, Potentiometric, NMR, and Crystallographic Studies. J. Med. Chem. 2018, 61, 4421–4435. [Google Scholar] [CrossRef]
- Li, Y.; Woster, P.M. Discovery of a new class of histone deacetylase inhibitors with a novel zinc binding group. MedChemComm 2015, 6, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Michel, M.; Homan, E.J.; Wiita, E.; Pedersen, K.; Almlöf, I.; Gustavsson, A.-L.; Lundbäck, T.; Helleday, T.; Berglund, U.W. In silico Druggability Assessment of the NUDIX Hydrolase Protein Family as a Workflow for Target Prioritization. Front. Chem. 2020, 8, 506842. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, S.; Cuffaro, D.; Nuti, E.; Ciccone, L.; Tuccinardi, T.; Liva, F.; D’andrea, F.; de Groot, R.; Rossello, A.; Ahnström, J. Exosite inhibition of ADAMTS-5 by a glycoconjugated arylsulfonamide. Sci. Rep. 2021, 11, 949. [Google Scholar] [CrossRef]
- Prieto-Martínez, F.D.; Arciniega, M.; Medina-Franco, J.L.; Prieto-Martínez, F.D.; Arciniega, M.; Medina-Franco, J.L. Molecular docking: Current advances and challenges. TIP Rev. Espec. En. Cienc. Químico Biológicas 2018, 21, 65–87. [Google Scholar] [CrossRef]
- Iqbal, M.; Kurniawan, R.V.; Nurfani, H.D.W.; Roestamadji, R.I.; Luthfi, M.; Setyowati, D.; Setijanto, R.D.; Surboyo, M.D.C. Molecular docking analysis of major active compounds of pomegranate peel extract (Punica granatum L.) in inhibiting cyclooxygenase enzyme. J. Adv. Res. Rev. 2023, 20, 1824–1842. [Google Scholar] [CrossRef]
- Shieh, H.S.; Tomasselli, A.G.; Mathis, K.J.; Schnute, M.E.; Woodard, S.S.; Caspers, N.; Williams, J.M.; Kiefer, J.R.; Munie, G.; Wittwer, A.; et al. Structure analysis reveals the flexibility of the ADAMTS-5 active site. Protein Sci. 2011, 20, 735–744. [Google Scholar] [CrossRef]
- Zentrum für Bioinformatik: Universität Hamburg-Proteins Plus Server. Available online: https://proteins.plus/ (accessed on 27 May 2024).
- Volkamer, A.; Kuhn, D.; Grombacher, T.; Rippmann, F.; Rarey, M. Combining global and local measures for structure-based druggability predictions. J. Chem. Inf. Model. 2012, 52, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys, Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Mehler, E.L.; Solmajer, T. Electrostatic effects in proteins: Comparison of dielectric and charge models. Protein Eng. 1991, 4, 903–910. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Binding Pocket | P1 | P2 | P3 |
|---|---|---|---|
 | 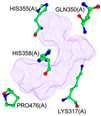 | 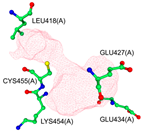 | |
| Volume (Å) | 728.9 | 468.1 | 212.3 |
| Surface (Å) | 815.7 | 1015.1 | 428 |
| Drug Binding Score | 0.77 | 0.72 | 0.53 |
| H Donors | 10 | 14 | 15 |
| H Acceptors | 32 | 40 | 38 |
| Hydrophobicity | 0.60 | 0.41 | 0.16 |
| Polar AA Ratio | 0.22 | 0.19 | 0.43 |
| Ligand | Volume (Å) | XLogP3 | PSA (Å) |
|---|---|---|---|
| EA | 221.78 | 1.1 | 141.33 |
| GA | 466.7 | 1.28 | 337.3 |
| PCG | 800.91 | 0.17 | 518.75 |
| HHDP | 257.56 | 0.47 | 195.97 |
| A2N | 419.64 | 2.61 * | 117.12 |
| Ligand | 3D Interaction | 2D Interaction | Pocket Fit |
|---|---|---|---|
| EA | 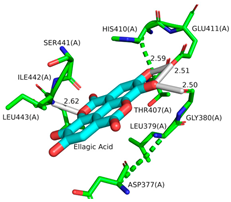 | 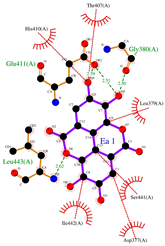 | 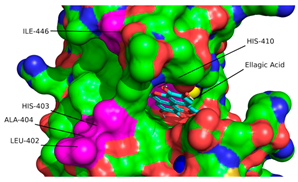 |
| PCG | 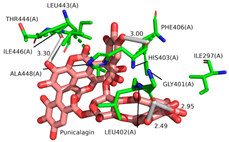 |  | 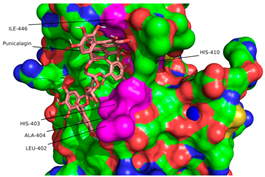 |
| GA | 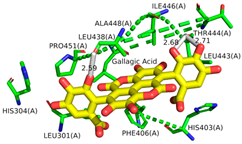 | 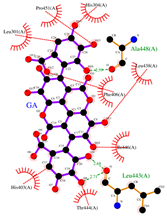 | 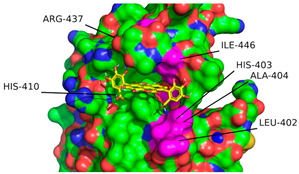 |
| HHDP | 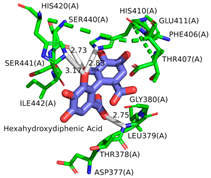 | 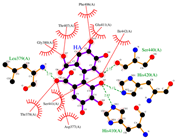 |  |
 | |||
| Sample | Area | Ratio (b/a) |
|---|---|---|
| a-1 | 52,658 | 1.00 |
| a-3 | 53,495 | 1.00 |
| a-6 | 60,182 | 1.00 |
| a-24 | 113,690 | 1.00 |
| b-1 | 96,634 | 1.84 |
| b-3 | 79,002 | 1.48 |
| b-6 | 72,104 | 1.20 |
| b-24 | 95,802 | 0.84 |
| Grid Box Inputs | |
|---|---|
| x-points | 60 |
| y-points | 60 |
| z-points | 60 |
| Spacing (Å) | 0.375 |
| x-center (Å) | 8.063 |
| y-center (Å) | 2.49 |
| z-center (Å) | 25.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breland, A.N.; Ross, M.K.; Fitzkee, N.C.; Elder, S.H. In Silico Insights into the Inhibition of ADAMTS-5 by Punicalagin and Ellagic Acid for the Treatment of Osteoarthritis. Int. J. Mol. Sci. 2025, 26, 4093. https://doi.org/10.3390/ijms26094093
Breland AN, Ross MK, Fitzkee NC, Elder SH. In Silico Insights into the Inhibition of ADAMTS-5 by Punicalagin and Ellagic Acid for the Treatment of Osteoarthritis. International Journal of Molecular Sciences. 2025; 26(9):4093. https://doi.org/10.3390/ijms26094093
Chicago/Turabian StyleBreland, Austen N., Matthew K. Ross, Nicholas C. Fitzkee, and Steven H. Elder. 2025. "In Silico Insights into the Inhibition of ADAMTS-5 by Punicalagin and Ellagic Acid for the Treatment of Osteoarthritis" International Journal of Molecular Sciences 26, no. 9: 4093. https://doi.org/10.3390/ijms26094093
APA StyleBreland, A. N., Ross, M. K., Fitzkee, N. C., & Elder, S. H. (2025). In Silico Insights into the Inhibition of ADAMTS-5 by Punicalagin and Ellagic Acid for the Treatment of Osteoarthritis. International Journal of Molecular Sciences, 26(9), 4093. https://doi.org/10.3390/ijms26094093