

Insights into Active Site Cysteine Residues in Mycobacterium tuberculosis Enzymes: Potential Targets for Anti-Tuberculosis Intervention




Abstract
1. Introduction
1.1. Mycobacterium tuberculosis and the Importance of Enzyme Function
1.2. Metalloenzymes as Potential Therapeutic Targets
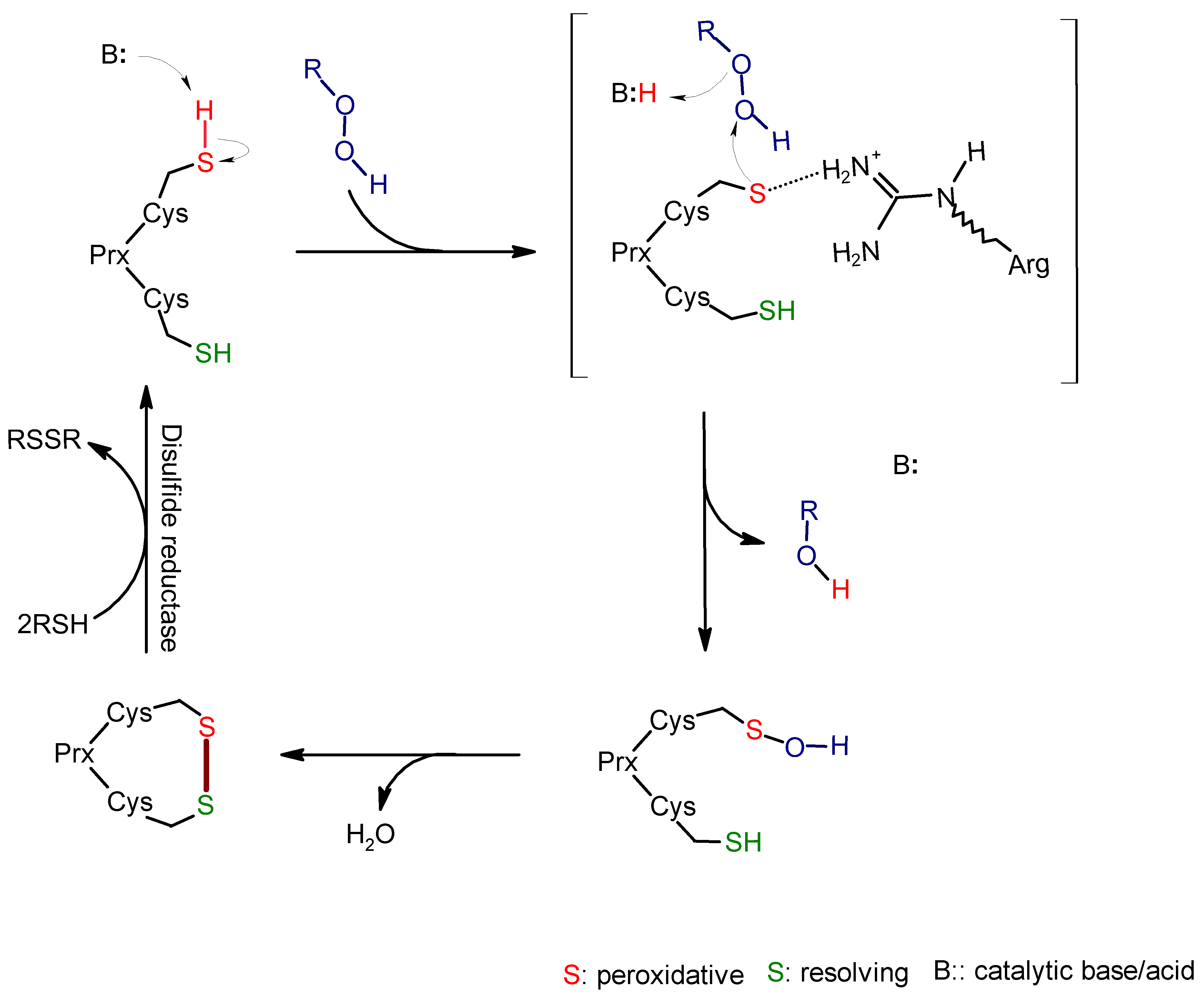
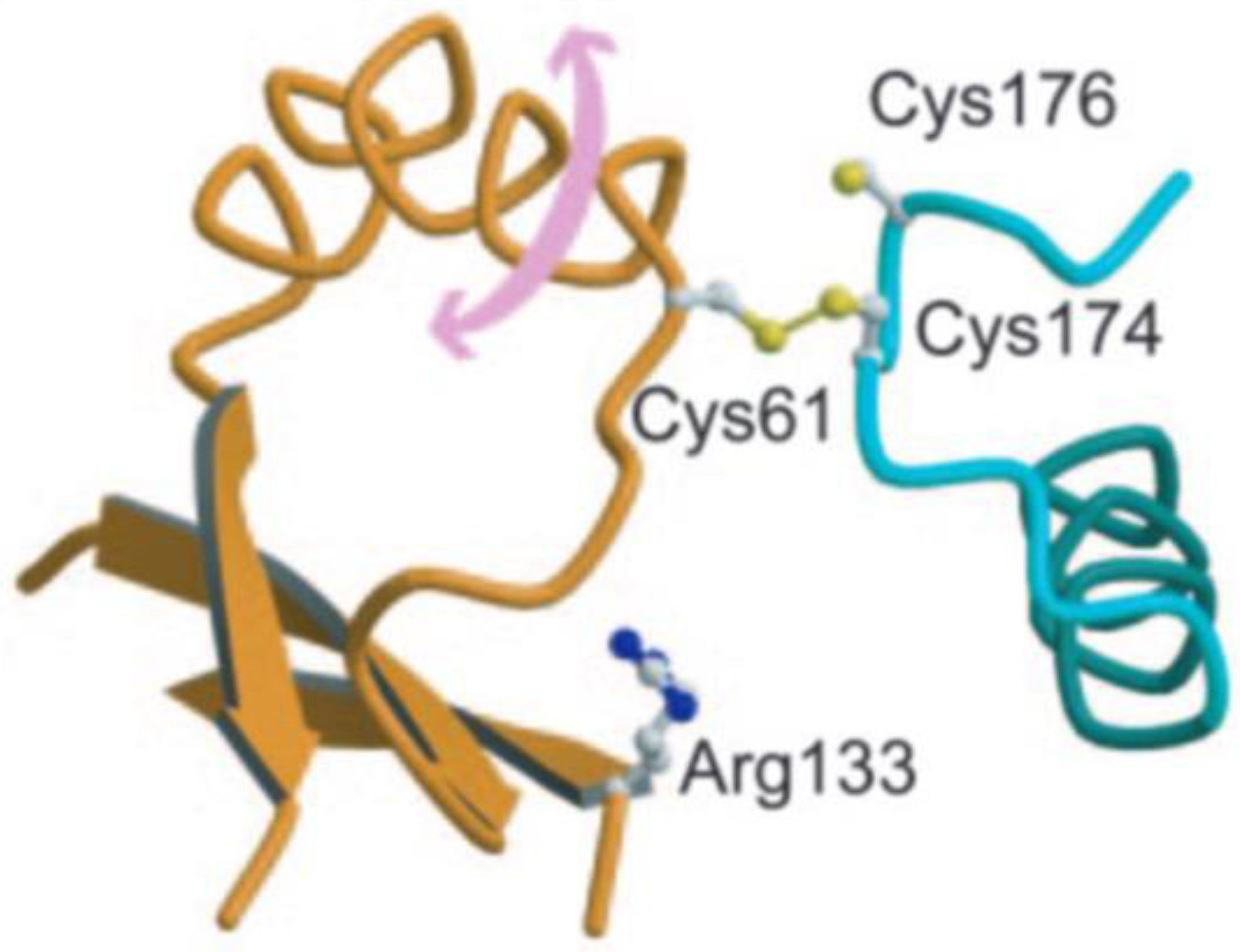
2. Alkyl Hydroperoxide Reductase C (AhpC)
2.1. Function and Significance of AhpC in M. tuberculosis
2.2. Role of Active Site Cysteine in AhpC Activity and Potential as a Drug Target
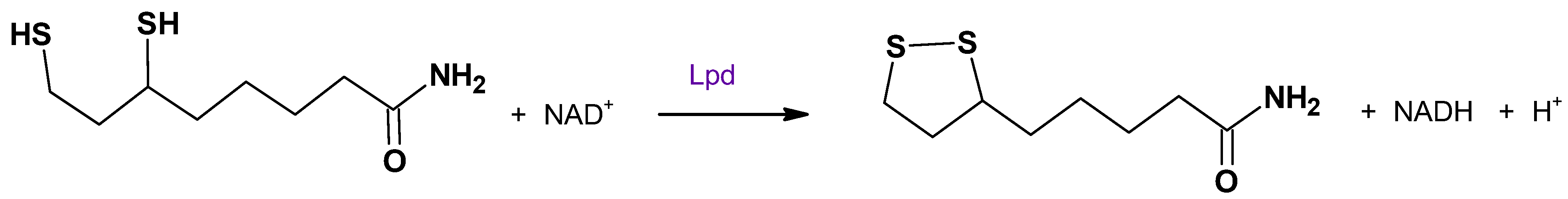
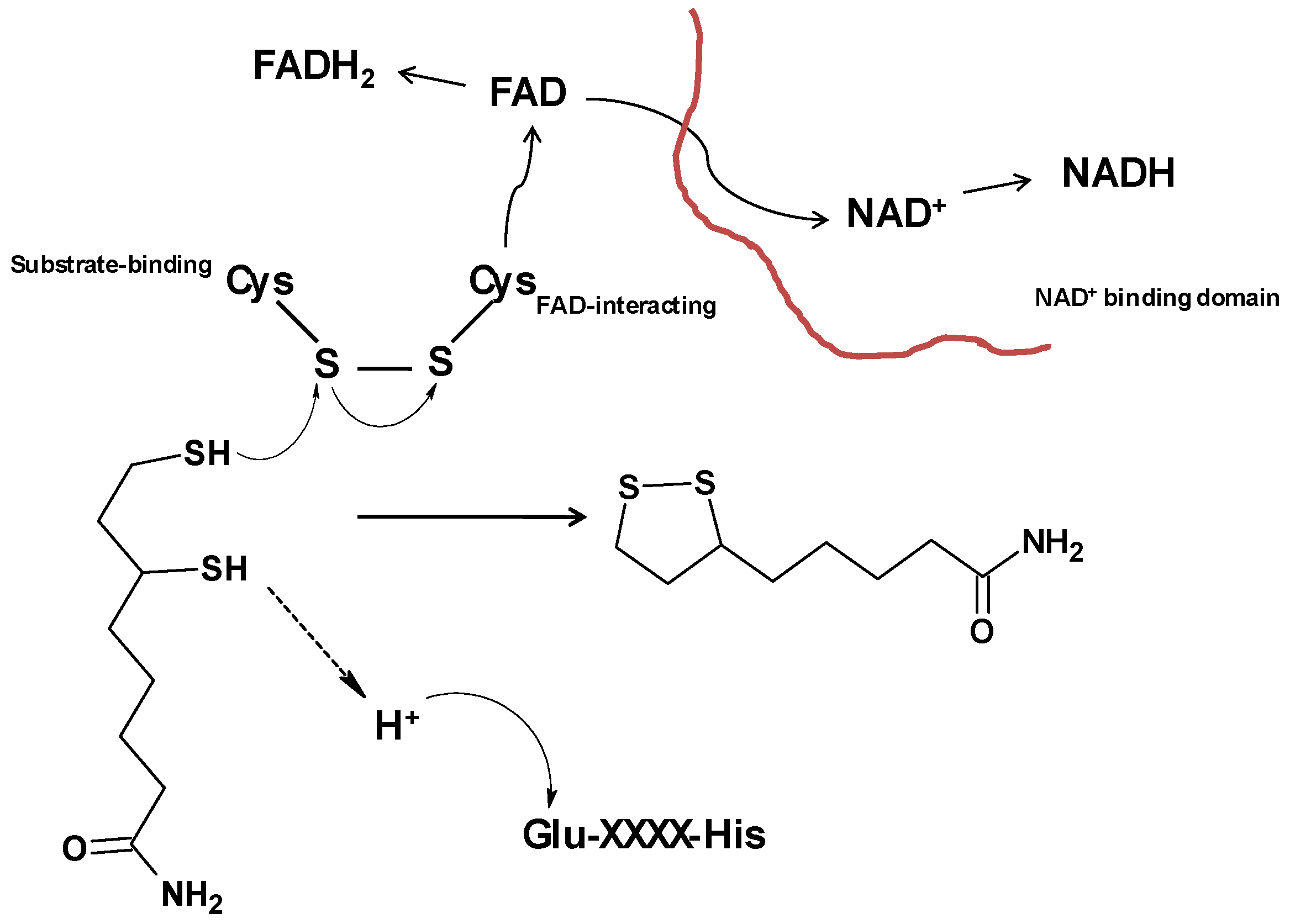
3. Dihydrolipoamide Dehydrogenase (Lpd)
3.1. Role of Lpd in M. tuberculosis Metabolism
3.2. Impact of Active Site Cysteine on Lpd Function and Drug Development
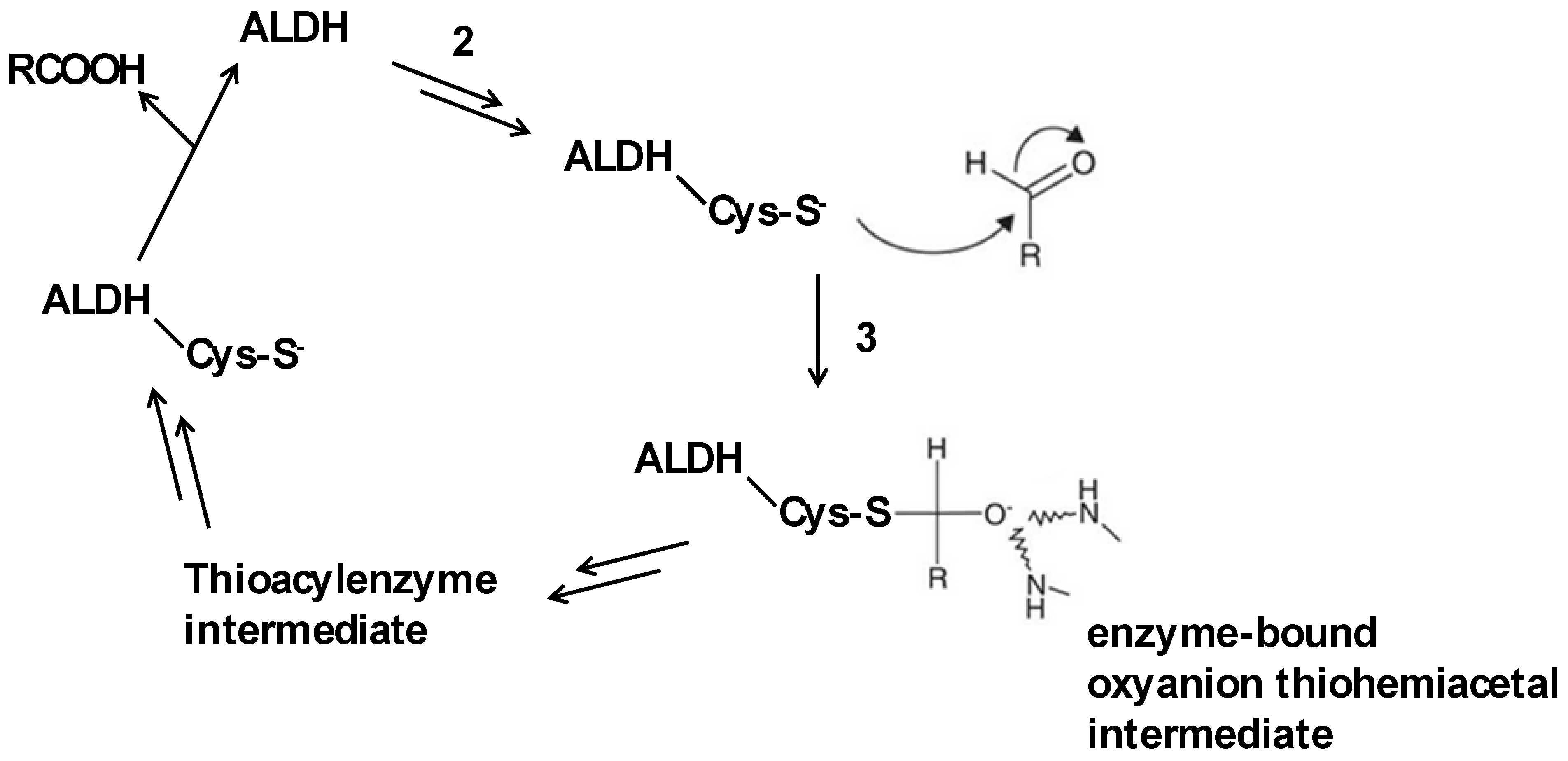
4. Aldehyde Dehydrogenase (ALDH)
4.1. Function of ALDH in Detoxification Pathways
4.2. Involvement of Active Site Cysteine in ALDH Activity and Drug Targeting Strategies
5. Methionine Aminopeptidase (MetAP)
5.1. Role of MetAP in Protein Processing
5.2. Significance of Active Site Cysteine in MetAP Function and Drug Targeting Strategies
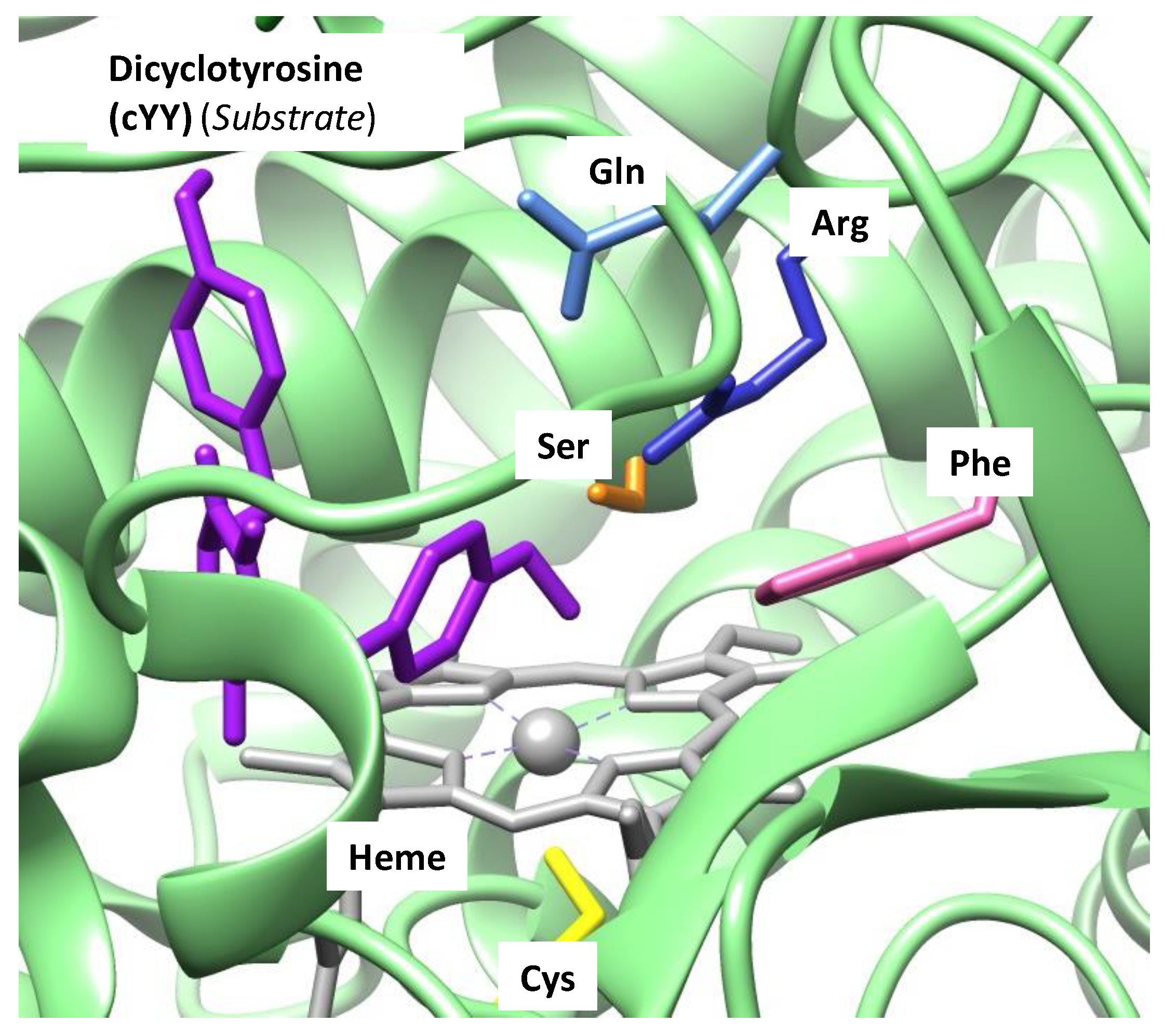
6. Cytochromes P450
7. Conclusions
7.1. Implications for Drug Discovery
7.2. Challenges and Future Research Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| TB | Tuberculosis |
| Mtb | Mycobacterium tuberculosis |
| AhpC | Alkyl hydroperoxide reductase |
| Lpd | Dihydrolipoamide dehydrogenase |
| ALDH | Aldehyde dehydrogenase |
| MetAP | Methionine aminopeptidase |
References
- Stokstad, E. Infectious disease: Drug-resistant TB on the rise. Science 2000, 287, 2391–2392. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). “Tuberculosis (TB)” 7 November 2023. Available online: https://www.who.int/news-room/fact-sheets/detail/tuberculosis (accessed on 24 October 2024).
- Chai, Q.; Zhang, Y.; Liu, C.H. Mycobacterium tuberculosis: An Adaptable Pathogen Associated with Multiple Human Diseases. Front. Microbiol. 2018, 8, 158. [Google Scholar] [CrossRef]
- Shah, M.; Chida, N. Extrapulmonary tuberculosis. In Handbook of Tuberculosis; Grosset, J.H., Chaisson, R.E., Eds.; Springer International Publishing: Cham, Switzerland, 2017; pp. 91–118. [Google Scholar]
- Chen, A.Y.; Adamek, R.N.; Dick, B.L.; Credille, C.V.; Morrison, C.N.; Cohen, S.M. Targeting Metalloenzymes for Therapeutic Intervention. Chem. Rev. 2019, 119, 1323–1455. [Google Scholar] [CrossRef]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-Containing Oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef]
- Meunier, B.; de Visser, S.P.; Shaik, S. Mechanism of Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005, 105, 2253–2277. [Google Scholar] [CrossRef]
- Munro, A.W.; Girvan, H.M.; McLean, K.J. Variations on a (T)Heme—Novel Mechanisms, Redox Partners and Catalytic Functions in the Cytochrome P450 Superfamily. Nat. Prod. Rep. 2007, 24, 585–609. [Google Scholar] [CrossRef]
- Green, M.T. C-H Bond Activation in Heme Proteins: The Role of Thiolate Ligation in Cytochrome P450. Curr. Opin. Chem. Biol. 2009, 13, 84–88. [Google Scholar] [CrossRef]
- Kadish, K.M.; Smith, K.M.; Guilard, R. (Eds.) Handbook of Porphyrin Science; World Scientific Publishing Co.: Hackensack, NJ, USA, 2010. [Google Scholar]
- Guengerich, F.P. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef]
- Dunham, N.P.; Arnold, F.H. Nature’s Machinery, Repurposed: Expanding the Repertoire of Iron-Dependent Oxygenases. ACS Catal. 2020, 10, 12239–12255. [Google Scholar] [CrossRef]
- de Visser, S.P.; Lin, Y.-T.; Ali, H.S.; Bagha, U.K.; Mukherjee, G.; Sastri, C.V. Negative Catalysis or Non-Bell-Evans-Polanyi Reactivity by Metalloenzymes: Examples from Mononuclear Heme and Non-Heme Iron Oxygenases. Coord. Chem. Rev. 2021, 439, 213914. [Google Scholar] [CrossRef]
- Zhang, Y.; Mokkawes, T.; de Visser, S.P. Insights into Cytochrome P450 Enzyme Catalyzed Defluorination of Aromatic Fluorides. Angew. Chem. Int. Ed. 2023, 62, e202310785. [Google Scholar] [CrossRef] [PubMed]
- Chapman, H.A.; Riese, R.J.; Shi, G.-P. Emerging roles for cysteine proteases in human biology. Annu. Rev. Physiol. 1997, 59, 63–88. [Google Scholar] [CrossRef] [PubMed]
- Otto, H.-H.; Schirmeister, T. Cysteine proteases and their inhibitors. Chem. Rev. 1997, 97, 133–171. [Google Scholar] [CrossRef]
- Madala, P.K.; Tyndall, J.D.A.; Nall, T.; Fairlie, D.P. Update 1 of: Proteases universally recognize beta strands in their active sites. Chem. Rev. 2010, 110, PR1–PR31. [Google Scholar] [CrossRef]
- Patil, N.A.; Tailhades, J.; Hughes, R.A.; Separovic, F.; Wade, J.D.; Hossain, M.A. Cellular disulfide bond formation in bioactive peptides and proteins. Int. J. Mol. Sci. 2015, 16, 1791–1805. [Google Scholar] [CrossRef]
- Bošnjak, I.; Bojović, V.; Šegvić-Bubić, T.; Bielen, A. Occurrence of protein disulfide bonds in different domains of life: A comparison of proteins from the Protein Data Bank. Protein Eng. Des. Sel. 2014, 27, 65–72. [Google Scholar] [CrossRef]
- Feige, M.J.; Braakman, I.; Hendershot, L.M. Oxidative Folding of Proteins: Basic Principles, Cellular Regulation and Engineering; Feige, M.J., Ed.; The Royal Society of Chemistry: London, UK, 2018; pp. 1–33. [Google Scholar]
- Fomenko, D.E.; Marino, S.M.; Gladyshev, V.N. Functional diversity of cysteine residues in proteins and unique features of catalytic redox-active cysteines in thiol oxidoreductases. Mol. Cell 2008, 26, 228–235. [Google Scholar] [CrossRef]
- Parish, T.; Stoker, N.G. The common aromatic amino acid biosynthesis pathway is essential in Mycobacterium tuberculosis. Microbiology 2002, 148, 3069–3077. [Google Scholar] [CrossRef]
- Roberts, F.; Roberts, C.W.; Johnson, J.J.; Kyle, D.E.; Krell, T.; Coggins, J.R.; Coombs, G.H.; Milhous, W.K.; Tzipori, S.; Ferguson, D.J.P.; et al. Evidence for the shikimate pathway in apicomplexan parasites. Nature 1998, 393, 801–805. [Google Scholar] [CrossRef]
- Campbell, S.A.; Richards, T.A.; Mui, E.J.; Samuel, B.U.; Coggins, J.R.; McLeod, R.; Roberts, C.W. A complete shikimate pathway in Toxoplasma gondii: An ancient eukaryotic innovation. Int. J. Parasitol. 2004, 34, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, K.M.; Weaver, L.M. The Shikimate Pathway. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 473–503. [Google Scholar] [CrossRef]
- Tzin, V.; Galili, G.; Aharoni, A. Shikimate Pathway and Aromatic Amino Acid Biosynthesis. eLS 2012, 1–10. [Google Scholar] [CrossRef]
- Faponle, A.S.; Fagbohunka, B.S.; Gauld, J.W. Influence of Cysteine440 on the Active Site Properties of 3-Deoxy-d-Arabino-Heptulosonate 7-Phosphate Synthase in Mycobacterium tuberculosis (MtDAHPS). ACS Omega 2023, 8, 14401–14409. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.F.; Shin, J.; Subramanian Manimekalai, M.S.; Saw, W.G.; Yin, Z.; Bhushan, S.; Kumar, A.; Ragunathan, P.; Grüber, G. AhpC of the mycobacterial antioxidant defense system and its interaction with its reducing partner Thioredoxin-C. Sci. Rep. 2017, 7, 5159. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xie, Q.W.; Nathan, C. Alkyl hydroperoxide reductase subunit C (AhpC) protects bacterial and human cells against reactive nitrogen intermediates. Mol. Cell 1998, 1, 795–805. [Google Scholar] [CrossRef]
- Bryk, R.; Lima, C.D.; Erdjument-Bromage, H.; Tempst, P.; Nathan, C. Metabolic enzymes of mycobacteria linked to antioxidant defense by a thioredoxin-like protein. Science 2002, 295, 1073–1077. [Google Scholar] [CrossRef]
- Bhargavi, G.; Singh, A.K.; Deenadayalan, A.; Ponnuraja, C.; Patil, S.A.; Palaniyandi, K. Role of a Putative Alkylhydroperoxidase Rv2159c in the Oxidative Stress Response and Virulence of Mycobacterium tuberculosis. Pathogens 2022, 11, 684. [Google Scholar] [CrossRef]
- Wilson, T.; de Lisle, G.W.; Marcinkeviciene, J.A.; Blanchardand, J.S.; Collins, D.M. Antisense RNA to ahpC, an oxidative stress defence gene involved in isoniazid resistance, indicates that AhpC of Mycobacterium bovis has virulence properties. Microbiology 1998, 144, 2687–2695. [Google Scholar] [CrossRef]
- Master, S.S.; Springer, B.; Sander, P.; Boettger, E.C.; Deretic, V.; Timmins, G.S. Oxidative stress response genes in Mycobacterium tuberculosis: Role of ahpC in resistance to peroxynitrite and stage-specific survival in macrophages. Microbiology 2002, 148, 3139–3144. [Google Scholar] [CrossRef]
- Wood, Z.A.; Schröder, E.; Robin Harris, J.; Poole, L.B. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem. Sci. 2003, 28, 32–40. [Google Scholar] [CrossRef]
- Guimarães, B.G.; Souchon, H.; Honoré, N.; Saint-Joanis, B.; Brosch, R.; Shepard, W.; Cole, S.T.; Alzari, P.M. Structure and mechanism of the alkyl hydroperoxidaseAhpC, a key element of the Mycobacterium tuberculosis defense system against oxidative stress. J. Biol. Chem. 2005, 280, 25735–25742. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, R.; Mande, S.C. Site-directed mutagenesis reveals a novel catalytic mechanism of Mycobacterium tuberculosis alkylhydroperoxidase C. Biochem. J. 2002, 367, 255–261. [Google Scholar] [CrossRef] [PubMed]
- Koshkin, A.; Knudsen, G.M.; Ortiz De Montellano, P.R. Intermolecular interactions in the AhpC/AhpD antioxidant defense system of Mycobacterium tuberculosis. Arch. Biochem. Biophys. 2004, 427, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Aluri, S.; de Visser, S.P. The mechanism of cysteine oxygenation by cysteine dioxygenase enzymes. J. Am. Chem. Soc. 2007, 129, 14846–14847. [Google Scholar] [CrossRef]
- Kumar, D.; Thiel, W.; de Visser, S.P. Theoretical study on the mechanism of the oxygen activation process in cysteine dioxygenase enzymes. J. Am. Chem. Soc. 2011, 133, 3869–3882. [Google Scholar] [CrossRef]
- Tchesnokov, E.P.; Faponle, A.S.; Davies, C.G.; Quesne, M.G.; Turner, R.; Fellner, M.; Souness, R.J.; Wilbanks, S.M.; de Visser, S.P.; Jameson, G.N.L. An iron-oxygen intermediate formed during the catalytic cycle of cysteine dioxygenase. Chem. Commun. 2016, 52, 8814–8817. [Google Scholar] [CrossRef]
- Faponle, A.S.; Seebeck, F.P.; de Visser, S.P. Sulfoxide synthase versus cysteine dioxygenase reactivity in a nonheme iron enzyme. J. Am. Chem. Soc. 2017, 139, 9259–9270. [Google Scholar] [CrossRef]
- Yeh, C.-C.G.; Pierides, C.; Jameson, G.N.L.; de Visser, S.P. Structure and functional differences of cysteine and 3-mercaptopropionate dioxygenases. A computational study. Chem. Eur. J. 2021, 27, 13793–13806. [Google Scholar] [CrossRef]
- Sherman, D.R.; Mdluli, K.; Hickey, M.J.; Arain, T.M.; Morris, S.L.; Barry, C.E.; Stover, C.K. Compensatory ahpC gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 1996, 272, 1641–1643. [Google Scholar] [CrossRef]
- Koshkin, A.; Zhou, X.T.; Kraus, C.N.; Brenner, J.M.; Bandyopadhyay, P.; Kuntz, I.D.; Barry, C.E.; Ortiz de Montellano, P.R. Inhibition of Mycobacterium tuberculosis AhpD, an element of the peroxiredoxin defense against oxidative stress. Antimicrob. Agents Chemother. 2004, 48, 2424–2430. [Google Scholar] [CrossRef] [PubMed]
- Carothers, D.J.; Pons, G.; Patel, M.S. Dihydrolipoamide dehydrogenase: Functional similarities and divergent evolution of the pyridine nucleotide-disulfide oxidoreductases. Arch. Biochem. Biophys. 1989, 268, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Bryk, R.; Itoh, M.; Suematsu, M.; Nathan, C. Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: Identification of alpha-ketoglutarate decarboxylase. Proc. Natl. Acad. Sci. USA 2005, 102, 10670–10675. [Google Scholar] [CrossRef] [PubMed]
- Argyrou, A.; Blanchard, J.S.; Palfey, B.A. The lipoamide dehydrogenase from Mycobacterium tuberculosis permits the direct observation of flavin intermediates in catalysis. Biochemistry 2002, 41, 14580–14590. [Google Scholar] [CrossRef]
- Rajashankar, K.R.; Bryk, R.; Kniewel, R.; Buglino, J.A.; Nathan, C.F.; Lima, C.D. Crystal structure and functional analysis of lipoamide dehydrogenase from Mycobacterium tuberculosis. J. Biol. Chem. 2005, 280, 33977–33983. [Google Scholar] [CrossRef]
- Venugopal, A.; Bryk, R.; Shi, S.; Rhee, K.; Rath, P.; Schnappinger, D.; Ehrt, S.; Nathan, C. Virulence of Mycobacterium tuberculosis depends on lipoamide dehydrogenase, a member of three multienzyme complexes. Cell Host Microbe 2011, 9, 21–31. [Google Scholar] [CrossRef]
- Hopkins, N.; Williams, C.H. Characterization of lipoamide dehydrogenase from Escherichia coli lacking the redox active disulfide: C44S and C49S. Biochemistry 1995, 34, 11757–11765. [Google Scholar] [CrossRef]
- Williams, C.H.J. Chemistry and Biochemistry of Flavoenzymes; Muller, F., Ed.; CRC Press: Boca Raton, FL, USA, 1992; pp. 121–212. [Google Scholar]
- Brautigam, C.A.; Chuang, J.L.; Tomchick, D.R.; Machius, M.; Chuang, D.T. Crystal structure of human dihydrolipoamide dehydrogenase: NAD+/NADH binding and the structural basis of disease-causing mutations. J. Mol. Biol. 2005, 350, 543–552. [Google Scholar] [CrossRef]
- Yan, L.J.; Wang, Y. Roles of Dihydrolipoamide Dehydrogenase in Health and Disease. Antioxid. Redox Signal. 2023, 39, 794–806. [Google Scholar] [CrossRef]
- Benen, J.; van Berkel, W.; Dieteren, N.; Arscott, D.; Williams, C.; Veeger, C.; de Kok, A. Lipoamide dehydrogenase from Azotobacter vinelandii: Site-directed mutagenesis of the His450-Glu455 diad. Kinetics of wild-type and mutated enzymes. Eur. J. Biochem. 1992, 207, 487–497. [Google Scholar] [CrossRef]
- Broxton, C.N.; Kaur, P.; Lavorato, M.; Ganesh, S.; Xiao, R.; Mathew, N.D.; Nakamaru-Ogiso, E.; Anderson, V.E.; Falk, M.J. Dichloroacetate and thiamine improve survival and mitochondrial stress in a C. elegans model of dihydrolipoamide dehydrogenase deficiency. JCI Insight 2022, 7, e156222. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, C.; Williams, C.H. Differential reactivity of the two active site cysteine residues generated on reduction of pig heart lipoamide dehydrogenase. J. Biol. Chem. 1976, 251, 3553–3557. [Google Scholar] [CrossRef]
- Becker, K.; Savvides, S.N.; Keese, M.; Schirmer, R.H.; Karplus, P.A. Enzyme inactivation through sulfhydryl oxidation by physiologic NO-carriers. Nat. Struct. Biol. 1998, 5, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Lipton, S.A. Redox modulation by S-nitrosylation contributes to protein misfolding, mitochondrial dynamics, and neuronal synaptic damage in neurodegenerative diseases. Cell Death Differ. 2011, 18, 1478–1486. [Google Scholar] [CrossRef]
- Yan, L.J.; Liu, L.; Forster, M.J. Reversible inactivation of dihydrolipoamide dehydrogenase by Angeli’s salt. Sheng Wu Wu Li Hsueh Bao 2012, 28, 341–350. [Google Scholar] [PubMed]
- Bryk, R.; Arango, N.; Venugopal, A.; Warren, J.D.; Park, Y.H.; Patel, M.S.; Lima, C.D.; Nathan, C. Triazaspirodimethoxybenzoyls as selective inhibitors of mycobacterial lipoamide dehydrogenase. Biochemistry 2010, 49, 1616–1627. [Google Scholar] [CrossRef]
- Kono, H.; Rusyn, I.; Yin, M.; Gäbele, E.; Yamashina, S.; Dikalova, A.; Kadiiska, M.B.; Connor, H.D.; Mason, R.P.; Segal, B.H.; et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J. Clin. Investig. 2000, 106, 867–872. [Google Scholar] [CrossRef]
- Szöcs, K.; Lassègue, B.; Wenzel, P.; Wendt, M.; Daiber, A.; Oelze, M.; Meinertz, T.; Münzel, T.; Baldus, S. Increased superoxide production in nitrate tolerance is associated with NAD(P)H oxidase and aldehyde dehydrogenase 2 downregulation. J. Mol. Cell. Cardiol. 2007, 42, 1111–1118. [Google Scholar] [CrossRef]
- Vasiliou, V.; Nebert, D.W. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Human Genomics 2005, 2, 138–143. [Google Scholar] [CrossRef]
- Marchitti, S.A.; Brocker, C.; Stagos, D.; Vasiliou, V. Non-P450 aldehyde oxidizing enzymes: The aldehyde dehydrogenase superfamily. Expert Opin. Drug Metab. Toxicol. 2008, 4, 697–720. [Google Scholar] [CrossRef]
- Kim, C.Y.; Webster, C.; Roberts, J.K.; Moon, J.H.; Alipio Lyon, E.Z.; Kim, H.; Yu, M.; Hung, L.W.; Terwilliger, T.C. Analysis of nucleoside-binding proteins by ligand-specific elution from dye resin: Application to Mycobacterium tuberculosis aldehyde dehydrogenases. J. Struct. Funct. Genom. 2009, 10, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Shortall, K.; Djeghader, A.; Magner, E.; Soulimane, T. Insights into Aldehyde Dehydrogenase Enzymes: A Structural Perspective. Front. Mol. Biosci. 2021, 8, 659550. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, J.; Stamler, J.S. Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 8306–8311. [Google Scholar] [CrossRef]
- Sydow, K.; Daiber, A.; Oelze, M.; Chen, Z.; August, M.; Wendt, M.; Ullrich, V.; Mülsch, A.; Schulz, E.; Keaney, J.F.; et al. Central role of mitochondrial aldehyde dehydrogenase and reactive oxygen species in nitroglycerin tolerance and cross-tolerance. J. Clin. Investig. 2004, 113, 482–489. [Google Scholar] [CrossRef]
- Cederbaum, A.I. Role of lipid peroxidation and oxidative stress in alcohol toxicity. Free Radical Biol. Med. 1989, 7, 537–539. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Zhou, J.; Hurley, T.D.; Weiner, H. Human liver mitochondrial aldehyde dehydrogenase: Three-dimensional structure and the restoration of solubility and activity of chimeric forms. Protein Sci. 1999, 8, 2784–2790. [Google Scholar] [CrossRef]
- Moon, J.H.; Lyon, A.E.; Yu, M.; Hung, L.-W.; Terwilliger, T.; Kim, C.-Y. X-ray Crystal Structure of Aldehyde Dehydrogenase from Mycobacterium Tuberculosis Complexed with NAD+ (PDB: 3B4W). Protein Data Bank. 2007. Available online: https://www.rcsb.org/structure/3B4W (accessed on 11 March 2025).
- Perez-Miller, S.J.; Hurley, T.D. Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase. Biochemistry 2003, 42, 7100–7109. [Google Scholar] [CrossRef]
- Nathan, C.; Shiloh, M.U. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc. Natl. Acad. Sci. USA 2000, 97, 8841–8848. [Google Scholar] [CrossRef]
- Wolschendorf, F.; Ackart, D.; Shrestha, T.B.; Hascall-Dove, L.; Nolan, S.; Lamichhane, G.; Wang, Y.; Bossmann, S.H.; Basaraba, R.J.; Niederweis, M. Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2011, 108, 1621–1626. [Google Scholar] [CrossRef]
- Darwin, K.H.; Stanley, S.A. The aldehyde hypothesis: Metabolic intermediates as antimicrobial effectors. Open Biol. 2022, 12, 220010. [Google Scholar] [CrossRef]
- Limón, G.; Samhadaneh, N.M.; Pironti, A.; Darwin, K.H. Aldehyde accumulation in Mycobacterium tuberculosis with defective proteasomal degradation results in copper sensitivity. mBio 2023, 14, e0036323. [Google Scholar] [CrossRef] [PubMed]
- Farrés, J.; Wang, T.T.; Cunningham, S.J.; Weiner, H. Investigation of the active site cysteine residue of rat liver mitochondrial aldehyde dehydrogenase by site-directed mutagenesis. Biochemistry 1995, 34, 2592–2598. [Google Scholar] [CrossRef]
- Steinmetz, C.G.; Xie, P.; Weiner, H.; Hurley, T.D. Structure of mitochondrial aldehyde dehydrogenase: The genetic component of ethanol aversion. Structure 1997, 5, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Horita, Y.; Takii, T.; Yagi, T.; Ogawa, K.; Fujiwara, N.; Inagaki, E.; Kremer, L.; Sato, Y.; Kuroishi, R.; Lee, Y.; et al. Antitubercular activity of disulfiram, an antialcoholism drug, against multidrug- and extensively drug-resistant Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 2012, 56, 4140–4145. [Google Scholar] [CrossRef] [PubMed]
- Dalecki, A.G.; Haeili, M.; Shah, S.; Speer, A.; Niederweis, M.; Kutsch, O.; Wolschendorf, F. Disulfiram and Copper Ions Kill Mycobacterium tuberculosis in a Synergistic Manner. Antimicrob. Agents Chemother. 2015, 59, 4835–4844. [Google Scholar] [CrossRef]
- Shen, M.L.; Lipsky, J.J.; Naylor, S. Role of disulfiram in the in vitro inhibition of rat liver mitochondrial aldehyde dehydrogenase. Biochem. Pharmacol. 2000, 60, 947–953. [Google Scholar] [CrossRef]
- Giglione, C.; Vallon, O.; Meinnel, T. Control of protein life-span by N-terminal methionine excision. EMBO J. 2003, 22, 13–23. [Google Scholar] [CrossRef]
- Lowther, W.T.; Matthews, B.W. Structure and function of the methionine aminopeptidases. Biochim. Biophys. Acta 2000, 1477, 157–167. [Google Scholar] [CrossRef]
- Griffith, E.C.; Su, Z.; Turk, B.E.; Chen, S.; Chang, Y.H.; Wu, Z.; Biemann, K.; Liu, J.O. Methionine aminopeptidase (type 2) is the common target for angiogenesis inhibitors AGM-1470 and ovalicin. Chem. Biol. 1997, 4, 461–471. [Google Scholar] [CrossRef]
- Ehlers, T.; Furness, S.; Robinson, T.P.; Zhong, H.A.; Goldsmith, D.; Aribser, J.; Bowen, J.P. Methionine AminoPeptidase Type-2 Inhibitors Targeting Angiogenesis. Curr. Top. Med. Chem. 2016, 16, 1478–1488. [Google Scholar] [CrossRef]
- Bernier, S.G.; Taghizadeh, N.; Thompson, C.D.; Westlin, W.F.; Hannig, G. Methionine aminopeptidases type I and type II are essential to control cell proliferation. J. Cell. Biochem. 2005, 95, 1191–1203. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Addlagatta, A.; Lu, J.; Matthews, B.W.; Liu, J.O. Elucidation of the function of type 1 human methionine aminopeptidase during cell cycle progression. Proc. Natl. Acad. Sci. USA 2006, 103, 18148–18153. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.Q.; Wang, J.J.; Zhang, C.M.; Liu, Z.P. The development of MetAP-2 inhibitors in cancer treatment. Curr. Med. Chem. 2012, 19, 1021–1035. [Google Scholar] [CrossRef]
- Arfin, S.M.; Kendall, R.L.; Hall, L.; Weaver, L.H.; Stewart, A.E.; Matthews, B.W.; Bradshaw, R.A. Eukaryotic methionyl aminopeptidases: Two classes of cobalt-dependent enzymes. Proc. Natl. Acad. Sci. USA 1995, 92, 7714–7718. [Google Scholar] [CrossRef]
- Bradshaw, R.A.; Brickey, W.W.; Walker, K.W. N-terminal processing: The methionine aminopeptidase and N alpha-acetyl transferase families. Trends Biochem. Sci. 1998, 23, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Addlagatta, A.; Quillin, M.L.; Omotoso, O.; Liu, J.O.; Matthews, B.W. Identification of an SH3-binding motif in a new class of methionine aminopeptidases from Mycobacterium tuberculosis suggests a mode of interaction with the ribosome. Biochemistry 2005, 44, 7166–7174. [Google Scholar] [CrossRef]
- Olaleye, O.; Raghunand, T.R.; Bhat, S.; He, J.; Tyagi, S.; Lamichhane, G.; Gu, P.; Zhou, J.; Zhang, Y.; Grosset, J.; et al. Methionine aminopeptidases from Mycobacterium tuberculosis as novel antimycobacterial targets. Chem. Biol. 2010, 17, 86–97. [Google Scholar] [CrossRef]
- Li, J.Y.; Cui, Y.M.; Chen, L.L.; Gu, M.; Li, J.; Nan, F.J.; Ye, Q.Z. Mutations at the S1 sites of methionine aminopeptidases from Escherichia coli and Homo sapiens reveal the residues critical for substrate specificity. J. Biol. Chem. 2004, 279, 21128–21134. [Google Scholar] [CrossRef]
- Swierczek, K.; Copik, A.J.; Swierczek, S.I.; Holz, R.C. Molecular discrimination of type-I over type-II methionyl aminopeptidases. Biochemistry 2005, 44, 12049–12056. [Google Scholar] [CrossRef]
- Reddi, R.; Arya, T.; Kishor, C.; Gumpena, R.; Ganji, R.J.; Bhukya, S.; Addlagatta, A. Selective targeting of the conserved active site cysteine of Mycobacterium tuberculosis methionine aminopeptidase with electrophilic reagents. FEBS J. 2014, 281, 4240–4248. [Google Scholar] [CrossRef]
- Chiu, C.H.; Lee, C.Z.; Lin, K.S.; Tam, M.F.; Lin, L.Y. Amino acid residues involved in the functional integrity of Escherichia coli methionine aminopeptidase. J. Bacteriol. 1999, 181, 4686–4689. [Google Scholar] [CrossRef] [PubMed]
- Lowther, W.T.; Matthews, B.W. Metalloaminopeptidases: Common functional themes in disparate structural surroundings. Chem. Rev. 2002, 102, 4581–4608. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. (Ed.) Cytochrome P450: Structure, Mechanism and Biochemistry, 3rd ed.; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2005. [Google Scholar]
- Krauser, J.A.; Guengerich, F.P. Cytochrome P450 3A4-Catalyzed Testosterone 6β-Hydroxylation Stereochemistry, Kinetic Deuterium Isotope Effects, and Rate-Limiting Steps. J. Biol. Chem. 2005, 280, 19496–19506. [Google Scholar] [CrossRef] [PubMed]
- Ortiz de Montellano, P.R. Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef]
- Spinello, A.; Pavlin, M.; Casalino, L.; Magistrato, A. A Dehydrogenase Dual Hydrogen Abstraction Mechanism Promotes Estrogen Biosynthesis: Can We Expand the Functional Annotation of the Aromatase Enzyme? Chem. Eur. J. 2018, 24, 10840–11849. [Google Scholar] [CrossRef]
- Huang, X.; Groves, J.T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef]
- Shaik, S.; de Visser, S.P.; Kumar, D. One Oxidant, Many Pathways: A Theoretical Perspective of Monooxygenation Mechanisms by Cytochrome P450 Enzymes. J. Biol. Inorg. Chem. 2004, 9, 661–668. [Google Scholar] [CrossRef] [PubMed]
- de Visser, S.P. Second-Coordination Sphere Effects on Selectivity and Specificity of Heme and Nonheme Iron Enzymes. Chem. Eur. J. 2020, 26, 5308–5327. [Google Scholar] [CrossRef]
- Nguyen, R.C.; Yang, Y.; Wang, Y.; Davis, I.; Liu, A. Substrate-Assisted Hydroxylation and O-Demethylation in the Peroxidase-like Cytochrome P450 Enzyme CYP121. ACS Catal. 2020, 10, 1628–1639. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Shaik, S.; Kumar, D.; de Visser, S.P.; Altun, A.; Thiel, W. Theoretical Perspective on the Structure and Mechanism of Cytochrome P450 Enzymes. Chem. Rev. 2005, 105, 2279–2328. [Google Scholar] [CrossRef]
- Rittle, J.; Green, M.T. Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science 2010, 330, 933–937. [Google Scholar] [CrossRef] [PubMed]
- Albertolle, M.E.; Kim, D.; Nagy, L.D.; Yun, C.H.; Pozzi, A.; Savas, Ü.; Johnson, E.F.; Guengerich, F.P. Heme-thiolate sulfenylation of human cytochrome P450 4A11 functions as a redox switch for catalytic inhibition. J. Biol. Chem. 2017, 292, 11230–11242. [Google Scholar] [CrossRef] [PubMed]
- Vatsis, K.P.; Peng, H.M.; Coon, M.J. Replacement of active-site cysteine-436 by serine converts cytochrome P450 2B4 into an NADPH oxidase with negligible monooxygenase activity. J. Inorg. Biochem. 2002, 91, 542–553. [Google Scholar] [CrossRef]
- İşci, Ü.; Faponle, A.S.; Afanasiev, P.; Albrieux, F.; Briois, V.; Ahsen, V.; Dumoulin, F.; Sorokin, A.B.; de Visser, S.P. Site-Selective Formation of an Iron(IV)-Oxo Species at the More Electron-Rich Iron Atom of Heteroleptic μ-Nitrido Diiron Phthalocyanines. Chem. Sci. 2015, 6, 5063–5075. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Inabe, K.; Shirakawa, Y.; Umezawa, N.; Kato, N.; Higuchi, T. Role of Thiolate Ligand in Spin State and Redox Switching in the Cytochrome P450 Catalytic Cycle. Inorg. Chem. 2017, 56, 4245–4248. [Google Scholar] [CrossRef]
- Ji, L.; Faponle, A.S.; Quesne, M.G.; Sainna, M.A.; Zhang, J.; Franke, A.; Kumar, D.; van Eldik, R.; Liu, W.; de Visser, S.P. Drug Metabolism by Cytochrome P450 Enzymes: What Distinguishes the Pathways Leading to Substrate Hydroxylation over Desaturation? Chem. Eur. J. 2015, 21, 9083–9092. [Google Scholar] [CrossRef]
- Faponle, A.S.; Quesne, M.G.; de Visser, S.P. Origin of the Regioselective Fatty Acid Hydroxylation versus Decarboxylation by a Cytochrome P450 Peroxygenase: What Drives the Reaction to Biofuel Production? Chem. Eur. J. 2016, 22, 5478–5483. [Google Scholar] [CrossRef]
- Li, X.-X.; Postils, V.; Sun, W.; Faponle, A.S.; Solà, M.; Wang, Y.; Nam, W.; de Visser, S.P. Reactivity Patterns of (Protonated) Compound II and Compound I of Cytochrome P450: Which is the Better Oxidant? Chem. Eur. J. 2017, 23, 6406–6418. [Google Scholar] [CrossRef]
- Qureshi, M.; Mokkawes, T.; Cao, Y.; de Visser, S.P. Mechanism of the Oxidative Ring-Closure Reaction During the Gliotoxin Biosynthesis by Cytochrome P450 GliF. Int. J. Mol. Sci. 2024, 25, 8567. [Google Scholar] [CrossRef]
- McLean, K.J.; Cheesman, M.R.; Rivers, S.L.; Richmond, A.; Leys, D.; Chapman, S.K.; Reid, G.A.; Price, N.C.; Kelly, S.M.; Clarkson, J.; et al. Expression, Purification and Spectroscopic Characterization of the Cy-tochrome P450 CYP121 from Mycobacterium tuberculosis. J. Inorg. Biochem. 2002, 91, 527–541. [Google Scholar] [CrossRef] [PubMed]
- McLean, K.J.; Carroll, P.; Lewis, D.G.; Dunford, A.J.; Seward, H.E.; Neeli, R.; Cheesman, M.R.; Marsollier, L.; Douglas, P.; Smith, W.E.; et al. Characterization of Active Site Structure in CYP121. A Cytochrome P450 Essential for Viability of Mycobacterium tuberculosis H37Rv. J. Biol. Chem. 2008, 283, 33406–33416. [Google Scholar] [CrossRef] [PubMed]
- Belin, P.; Le Du, M.H.; Fielding, A.; Lequin, O.; Jacquet, M.; Charbonnier, J.B.; Lecoq, A.; Thai, R.; Courçon, M.; Masson, C.; et al. Identification and Structural Basis of the Reaction Catalyzed by CYP121, an Essential Cytochrome P450 in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2009, 106, 7426–7431. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faponle, A.S.; Gauld, J.W.; de Visser, S.P. Insights into Active Site Cysteine Residues in Mycobacterium tuberculosis Enzymes: Potential Targets for Anti-Tuberculosis Intervention. Int. J. Mol. Sci. 2025, 26, 3845. https://doi.org/10.3390/ijms26083845
Faponle AS, Gauld JW, de Visser SP. Insights into Active Site Cysteine Residues in Mycobacterium tuberculosis Enzymes: Potential Targets for Anti-Tuberculosis Intervention. International Journal of Molecular Sciences. 2025; 26(8):3845. https://doi.org/10.3390/ijms26083845
Chicago/Turabian StyleFaponle, Abayomi S., James W. Gauld, and Sam P. de Visser. 2025. "Insights into Active Site Cysteine Residues in Mycobacterium tuberculosis Enzymes: Potential Targets for Anti-Tuberculosis Intervention" International Journal of Molecular Sciences 26, no. 8: 3845. https://doi.org/10.3390/ijms26083845
APA StyleFaponle, A. S., Gauld, J. W., & de Visser, S. P. (2025). Insights into Active Site Cysteine Residues in Mycobacterium tuberculosis Enzymes: Potential Targets for Anti-Tuberculosis Intervention. International Journal of Molecular Sciences, 26(8), 3845. https://doi.org/10.3390/ijms26083845