Proteome Alterations in Cardiac Fibroblasts: Insights from Experimental Myocardial Infarction and Clinical Ischaemic Cardiomyopathy

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
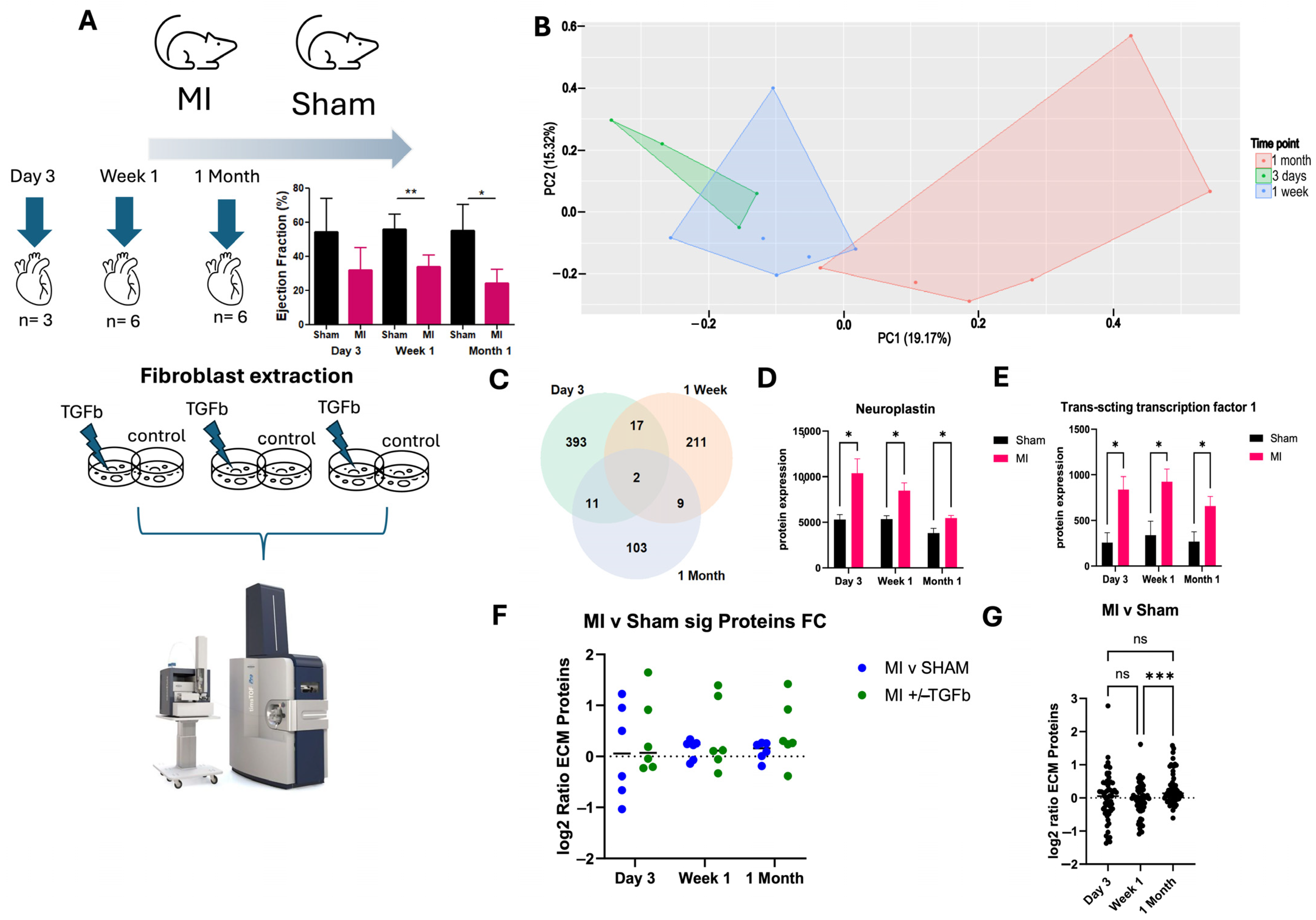
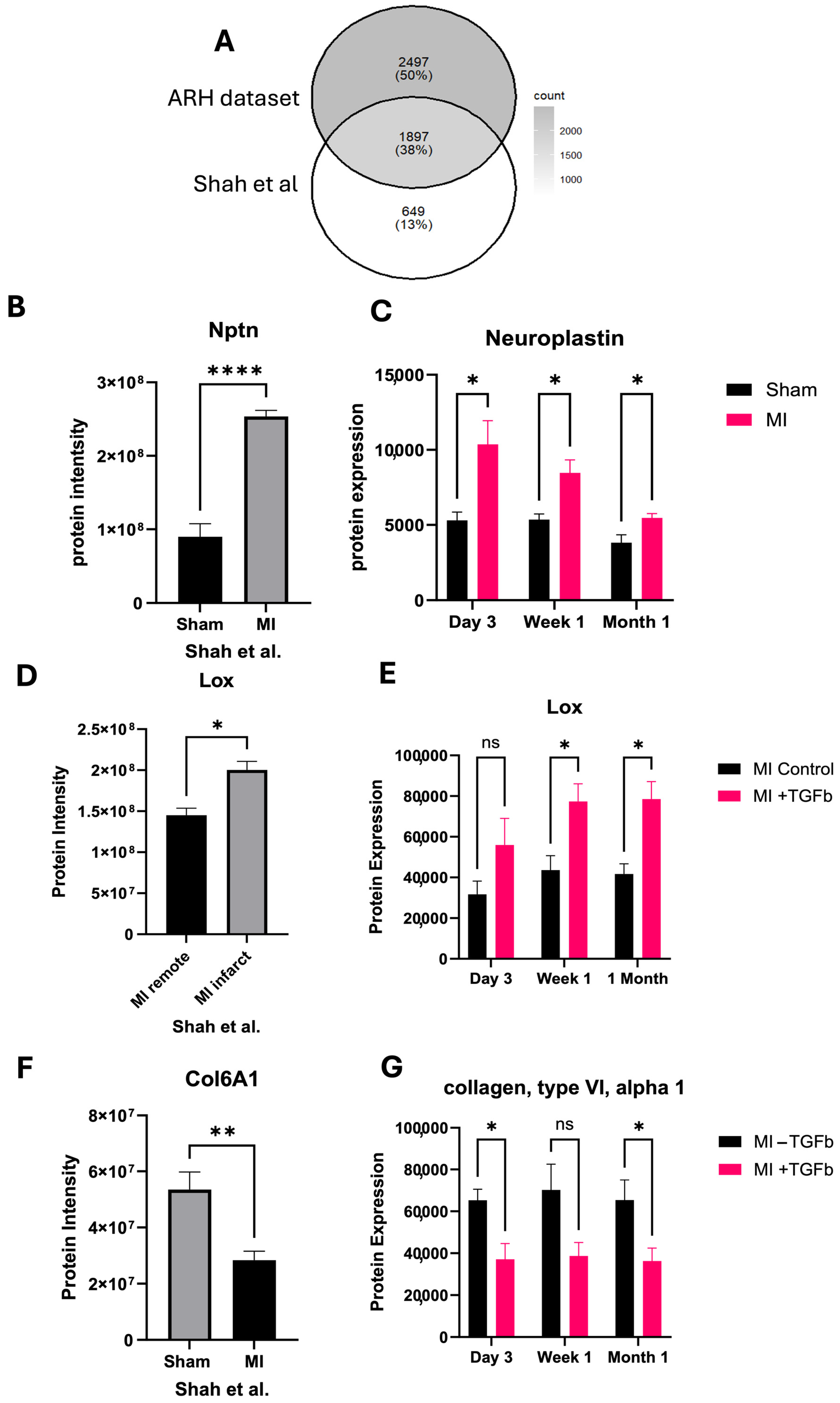
2.1. Proteomic Characterisation of Myocardial Infarction Reveals Unique Proteomic Profile of Chronic Injury
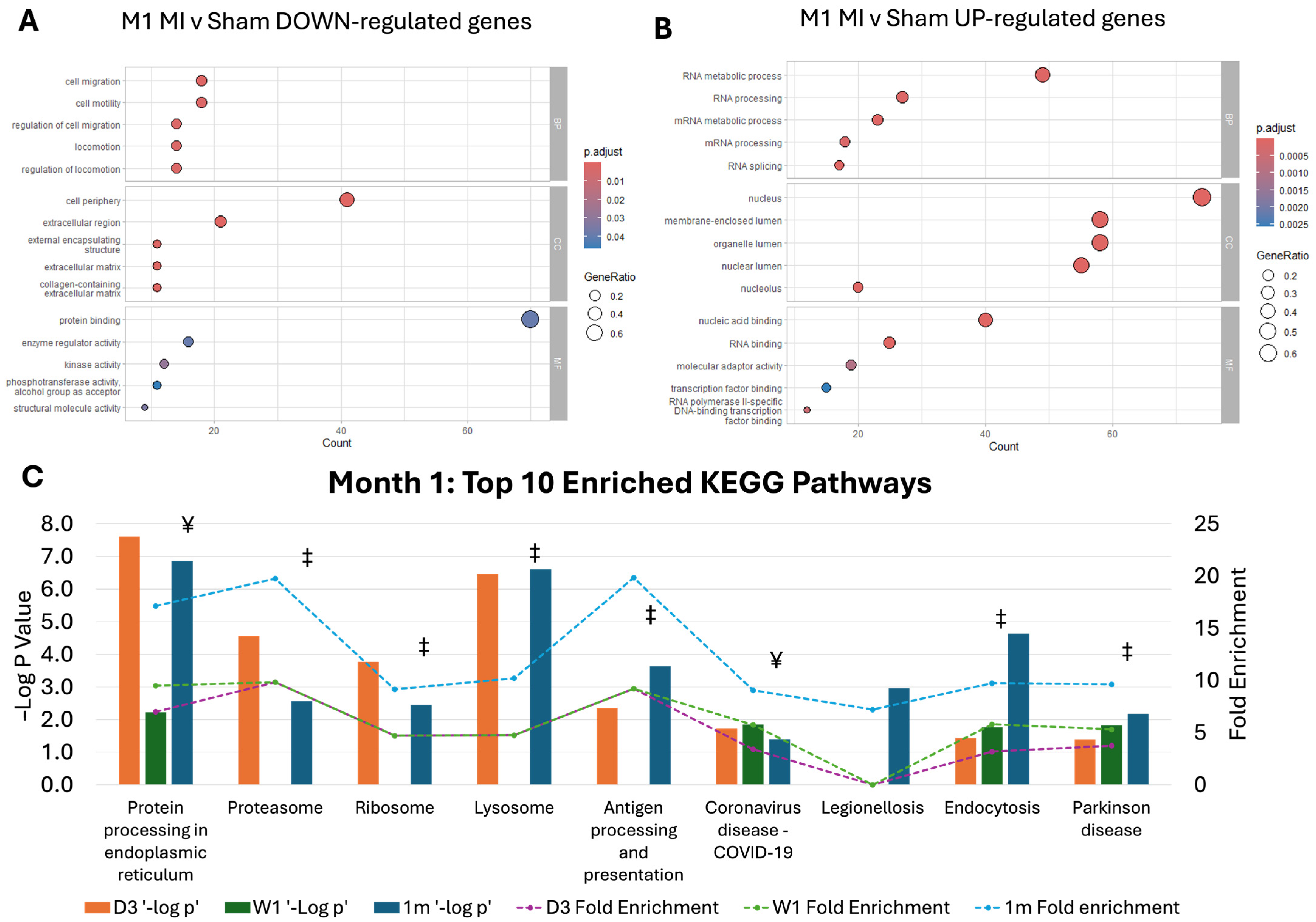
2.2. Pathway Dysregulation in Chronic MI Model
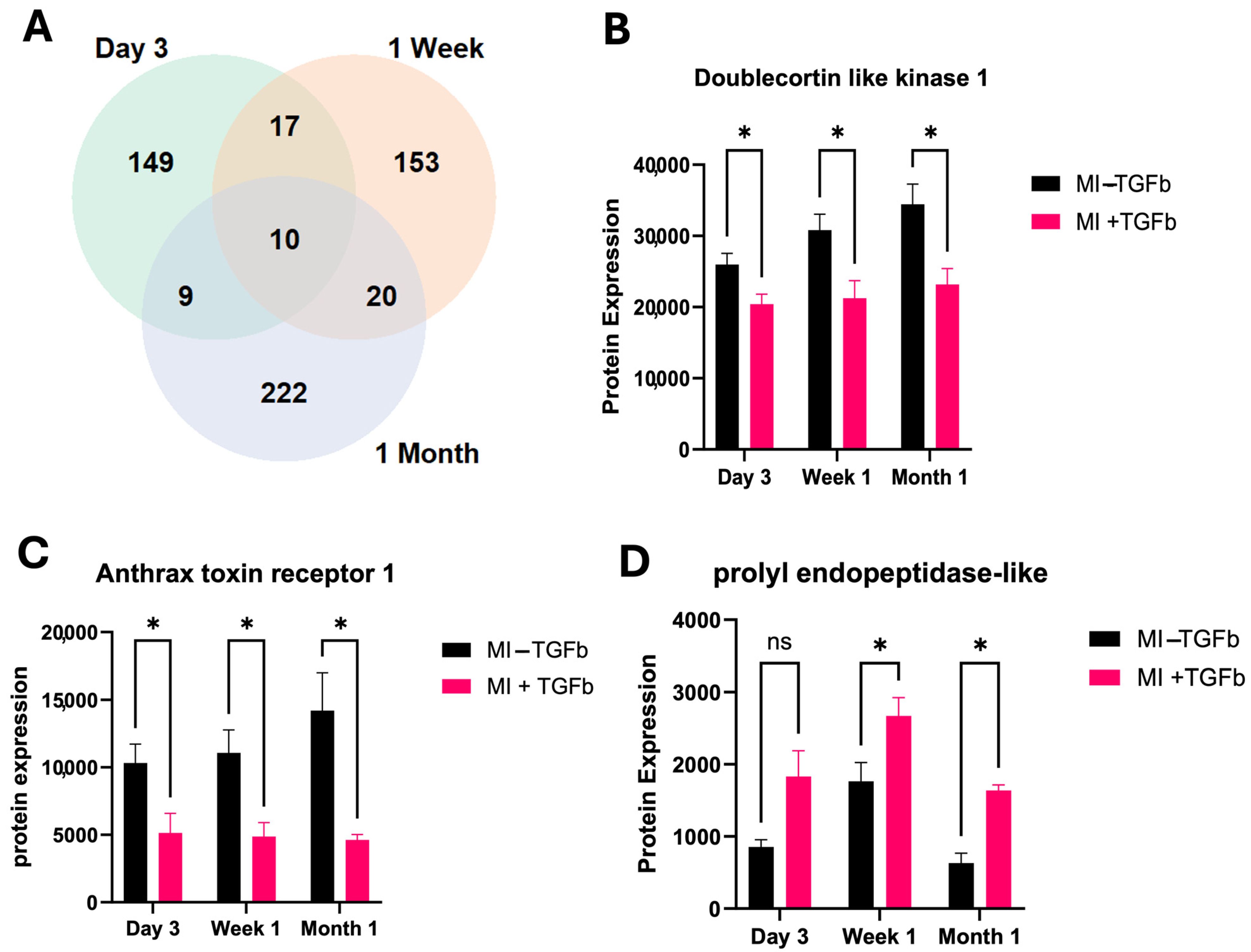
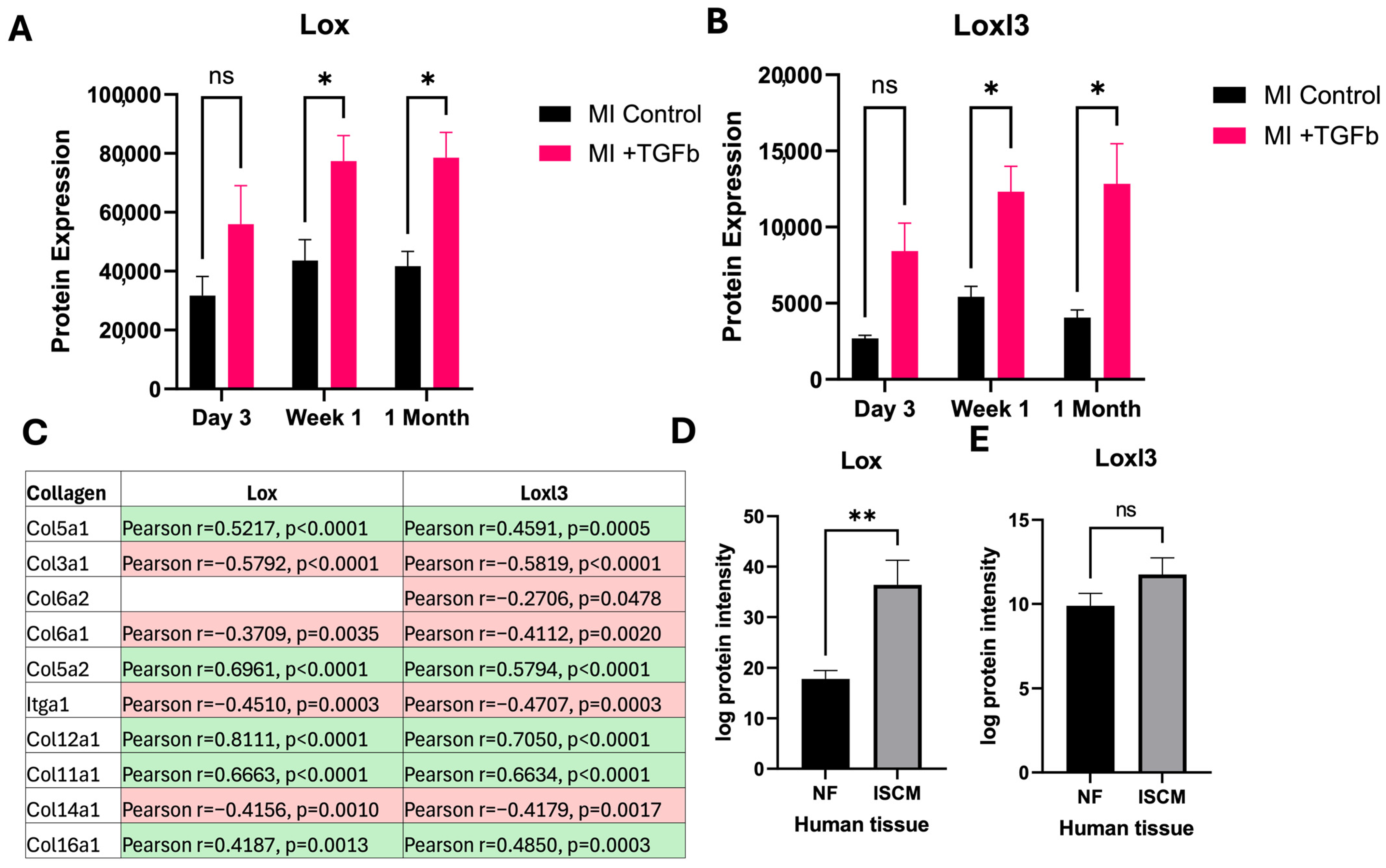
2.3. Effect of TGF-β Treatment on MI Cardiac Fibroblasts
2.4. In Silico Validation of Proteomic Changes in Cardiac Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Human Data and Analysis
4.2. In Vivo Model of Experimental MI
4.3. Echocardiography Data Acquisition and Data Analysis
4.4. Cardiac Fibroblast Isolation and Culture
4.5. Sample Preparation for Mass Spectrometry Analysis
4.6. Mass Spectrometry Analysis
4.7. Data Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IHD | ischaemic heart disease |
| HF | heart failure |
| TGF-β | Transcription Growth Factor Beta |
| LC-MS/MS | Liquid Chromatography Tandem Mass Spectrometry |
| ISCM | ischaemic cardiomyopathy |
| NF | Non-failure |
| MI | myocardial infarction |
| ECM | extracellular matrix |
| FDA | Food and Drug Administration |
| LV | left ventricular |
| MS | mass spectrometry |
| LAD | left anterior descending |
| UK | United Kingdom |
| BPM | Beats per minute |
| ECG | Electrocardiogram |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FBS | fetal bovine serum |
| BCA | Bicinchoninic acid assay |
| DTT | 1.4-dithiothreitol |
| IAA | iodoacetamide |
| TFA | trifluoroacetic acid |
| AcN | acetonitrile |
| DDA | Data dependent acquisition |
| DIA | Data independent acquisition |
| TIMS | trapped ion mobility spectrometry |
| PASEF | Parallel accumulation serial fragmentation |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| RNA | Ribonucleic acid |
| Dclk1 | doublecortin-like kinase 1 |
| Antxr1 | anthrax toxin receptor 1 |
| Prepl | prolyl endopeptidase-like protein |
| Lox | protein-lysine 6-oxidase |
| Loxl3 | Lysyl oxidase homolog 3 |
| Col6a1 | Protein collagen, type VI, alpha 1 |
| ER | Endoplasmic reticulum |
| NF-kB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
References
- Venugopal, H.; Hanna, A.; Humeres, C.; Frangogiannis, N.G. Properties and Functions of Fibroblasts and Myofibroblasts in Myocardial Infarction. Cells 2022, 11, 1386. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yan, J.; Hua, Z.; Jia, J.; Zhou, Z.; Zhang, J.; Li, J.; Zhang, J. Identification of molecular signatures in acute myocardial infarction based on integrative analysis of proteomics and transcriptomics. Genomics 2023, 115, 110701. [Google Scholar] [CrossRef] [PubMed]
- Kanderian, A.S.; Francis, G.S. Cardiac troponins and chronic kidney disease. Kidney Int. 2006, 69, 1112–1114. [Google Scholar] [CrossRef]
- Sun, Z.; Yun, Z.; Lin, J.; Sun, X.; Wang, Q.; Duan, J.; Li, C.; Zhang, X.; Xu, S.; Wang, Z.; et al. Comprehensive mendelian randomization analysis of plasma proteomics to identify new therapeutic targets for the treatment of coronary heart disease and myocardial infarction. J. Transl. Med. 2024, 22, 404. [Google Scholar] [CrossRef]
- Masarie, F.E., Jr.; Miller, R.A. Medical Subject Headings and medical terminology: An analysis of terminology used in hospital charts. Bull. Med. Libr. Assoc. 1987, 75, 89–94. [Google Scholar]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Tani, H.; Sadahiro, T.; Yamada, Y.; Isomi, M.; Yamakawa, H.; Fujita, R.; Abe, Y.; Akiyama, T.; Nakano, K.; Kuze, Y.; et al. Direct Reprogramming Improves Cardiac Function and Reverses Fibrosis in Chronic Myocardial Infarction. Circulation 2023, 147, 223–238. [Google Scholar] [CrossRef]
- Ko, T.; Nomura, S.; Yamada, S.; Fujita, K.; Fujita, T.; Satoh, M.; Oka, C.; Katoh, M.; Ito, M.; Katagiri, M.; et al. Cardiac fibroblasts regulate the development of heart failure via Htra3-TGF-beta-IGFBP7 axis. Nat. Commun. 2022, 13, 3275. [Google Scholar] [CrossRef]
- Shah, H.; Hacker, A.; Langburt, D.; Dewar, M.; McFadden, M.J.; Zhang, H.; Kuzmanov, U.; Zhou, Y.Q.; Hussain, B.; Ehsan, F.; et al. Myocardial Infarction Induces Cardiac Fibroblast Transformation within Injured and Noninjured Regions of the Mouse Heart. J. Proteome Res. 2021, 20, 2867–2881. [Google Scholar] [CrossRef]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef]
- Conrad, N.; Judge, A.; Tran, J.; Mohseni, H.; Hedgecott, D.; Crespillo, A.P.; Allison, M.; Hemingway, H.; Cleland, J.G.; McMurray, J.J.V.; et al. Temporal trends and patterns in heart failure incidence: A population-based study of 4 million individuals. Lancet 2018, 391, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.S.; Aday, A.W.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Barone Gibbs, B.; Beaton, A.Z.; Boehme, A.K.; et al. 2024 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation 2024, 149, e347–e913. [Google Scholar] [PubMed]
- Frantz, S.; Hundertmark, M.J.; Schulz-Menger, J.; Bengel, F.M.; Bauersachs, J. Left ventricular remodelling post-myocardial infarction: Pathophysiology, imaging, and novel therapies. Eur. Heart J. 2022, 43, 2549–2561. [Google Scholar] [CrossRef]
- Madsen, L.H.; Christensen, G.; Lund, T.; Serebruany, V.L.; Granger, C.B.; Hoen, I.; Grieg, Z.; Alexander, J.H.; Jaffe, A.S.; Van Eyk, J.E.; et al. Time course of degradation of cardiac troponin I in patients with acute ST-elevation myocardial infarction: The ASSENT-2 troponin substudy. Circ. Res. 2006, 99, 1141–1147. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, L.; Wang, S.; Cheng, H.; Xu, L.; Pei, G.; Wang, Y.; Fu, C.; Jiang, Y.; He, C.; et al. Signaling pathways and targeted therapy for myocardial infarction. Signal Transduct. Target. Ther. 2022, 7, 78. [Google Scholar] [CrossRef]
- Sun, Y. Myocardial repair/remodelling following infarction: Roles of local factors. Cardiovasc. Res. 2009, 81, 482–490. [Google Scholar] [CrossRef]
- Yagi, T.; Asada, R.; Kanekura, K.; Eesmaa, A.; Lindahl, M.; Saarma, M.; Urano, F. Neuroplastin Modulates Anti-inflammatory Effects of MANF. iScience 2020, 23, 101810. [Google Scholar] [CrossRef]
- Xie, R.; Zhang, Y.; Zhang, J.; Li, J.; Zhou, X. The Role of Circular RNAs in Immune-Related Diseases. Front. Immunol. 2020, 11, 545. [Google Scholar] [CrossRef]
- Yu, S.; Sun, Z.; Ju, T.; Liu, Y.; Mei, Z.; Wang, C.; Qu, Z.; Li, N.; Wu, F.; Liu, K.; et al. The m7G Methyltransferase Mettl1 Drives Cardiac Hypertrophy by Regulating SRSF9-Mediated Splicing of NFATc4. Adv. Sci. 2024, 11, e2308769. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Daseke, M.J., 2nd; Tenkorang, M.A.A.; Chalise, U.; Konfrst, S.R.; Lindsey, M.L. Cardiac fibroblast activation during myocardial infarction wound healing: Fibroblast polarization after MI. Matrix Biol. 2020, 91–92, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zou, Y.; Song, C.; Cao, K.; Cai, K.; Chen, S.; Zhang, Z.; Geng, D.; Zhang, N.; Feng, H.; et al. The role of serine/threonine protein kinases in cardiovascular disease and potential therapeutic methods. Biomed. Pharmacother. 2024, 177, 117093. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zhao, Y.; Luo, W.; Zhu, W.; Jin, L.; Wang, M.; Ye, L.; Wang, Y.; Liang, G. Macrophage DCLK1 promotes obesity-induced cardiomyopathy via activating RIP2/TAK1 signaling pathway. Cell Death Dis. 2023, 14, 419. [Google Scholar] [CrossRef]
- Huang, Z.; Shen, S.; Han, X.; Li, W.; Luo, W.; Lin, L.; Xu, M.; Wang, Y.; Huang, W.; Wu, G.; et al. Macrophage DCLK1 promotes atherosclerosis via binding to IKKbeta and inducing inflammatory responses. EMBO Mol. Med. 2023, 15, e17198. [Google Scholar] [CrossRef]
- Ji, L.; Yang, X.; Jin, Y.; Li, L.; Yang, B.; Zhu, W.; Xu, M.; Wang, Y.; Wu, G.; Luo, W.; et al. Blockage of DCLK1 in cardiomyocytes suppresses myocardial inflammation and alleviates diabetic cardiomyopathy in streptozotocin-induced diabetic mice. Biochim. Biophys. Acta Mol. Basis Dis. 2024, 1870, 166900. [Google Scholar] [CrossRef]
- Besschetnova, T.Y.; Ichimura, T.; Katebi, N.; St Croix, B.; Bonventre, J.V.; Olsen, B.R. Regulatory mechanisms of anthrax toxin receptor 1-dependent vascular and connective tissue homeostasis. Matrix Biol. 2015, 42, 56–73. [Google Scholar] [CrossRef]
- Przyklenk, M.; Karmacharya, S.; Bonasera, D.; Pasanen-Zentz, A.L.; Kmoch, S.; Paulsson, M.; Wagener, R.; Liccardi, G.; Schiavinato, A. ANTXR1 deficiency promotes fibroblast senescence: Implications for GAPO syndrome as a progeroid disorder. Sci. Rep. 2024, 14, 9321. [Google Scholar] [CrossRef]
- Ramachandra, C.J.A.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.H.; Hausenloy, D.J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef]
- Rosier, K.; McDevitt, M.T.; Smet, J.; Floyd, B.J.; Verschoore, M.; Marcaida, M.J.; Bingman, C.A.; Lemmens, I.; Dal Peraro, M.; Tavernier, J.; et al. Prolyl endopeptidase-like is a (thio)esterase involved in mitochondrial respiratory chain function. iScience 2021, 24, 103460. [Google Scholar] [CrossRef]
- Kagan, H.M.; Trackman, P.C. Properties and function of lysyl oxidase. Am. J. Respir. Cell Mol. Biol. 1991, 5, 206–210. [Google Scholar] [CrossRef]
- Rodriguez, C.; Martinez-Gonzalez, J. The Role of Lysyl Oxidase Enzymes in Cardiac Function and Remodeling. Cells 2019, 8, 1483. [Google Scholar] [CrossRef] [PubMed]
- Lopez, B.; Gonzalez, A.; Hermida, N.; Valencia, F.; de Teresa, E.; Diez, J. Role of lysyl oxidase in myocardial fibrosis: From basic science to clinical aspects. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1–H9. [Google Scholar] [CrossRef] [PubMed]
- Heymans, S.; Gonzalez, A.; Pizard, A.; Papageorgiou, A.P.; Lopez-Andres, N.; Jaisser, F.; Thum, T.; Zannad, F.; Diez, J. Searching for new mechanisms of myocardial fibrosis with diagnostic and/or therapeutic potential. Eur. J. Heart Fail. 2015, 17, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Martinez-Gonzalez, J.; Raposo, B.; Alcudia, J.F.; Guadall, A.; Badimon, L. Regulation of lysyl oxidase in vascular cells: Lysyl oxidase as a new player in cardiovascular diseases. Cardiovasc. Res. 2008, 79, 7–13. [Google Scholar] [CrossRef]
- Schiopu, A.; Bjorkbacka, H.; Narasimhan, G.; Loong, B.J.; Engstrom, G.; Melander, O.; Orho-Melander, M.; Nilsson, J. Elevated soluble LOX-1 predicts risk of first-time myocardial infarction. Ann. Med. 2023, 55, 2296552. [Google Scholar] [CrossRef]
- Gonzalez-Santamaria, J.; Villalba, M.; Busnadiego, O.; Lopez-Olaneta, M.M.; Sandoval, P.; Snabel, J.; Lopez-Cabrera, M.; Erler, J.T.; Hanemaaijer, R.; Lara-Pezzi, E.; et al. Matrix cross-linking lysyl oxidases are induced in response to myocardial infarction and promote cardiac dysfunction. Cardiovasc. Res. 2016, 109, 67–78. [Google Scholar] [CrossRef]
- Crossman, D.J.; Shen, X.; Jullig, M.; Munro, M.; Hou, Y.; Middleditch, M.; Shrestha, D.; Li, A.; Lal, S.; Dos Remedios, C.G.; et al. Increased collagen within the transverse tubules in human heart failure. Cardiovasc. Res. 2017, 113, 879–891. [Google Scholar] [CrossRef]
- Bryant, J.E.; Shamhart, P.E.; Luther, D.J.; Olson, E.R.; Koshy, J.C.; Costic, D.J.; Mohile, M.V.; Dockry, M.; Doane, K.J.; Meszaros, J.G. Cardiac myofibroblast differentiation is attenuated by α3 integrin blockade: Potential role in post-MI remodeling. J. Mol. Cell. Cardiol. 2009, 46, 186–192. [Google Scholar] [CrossRef]
- Naugle, J.E.; Olson, E.R.; Zhang, X.; Mase, S.E.; Pilati, C.F.; Maron, M.B.; Folkesson, H.G.; Horne, W.I.; Doane, K.J.; Meszaros, J.G. Type VI collagen induces cardiac myofibroblast differentiation: Implications for postinfarction remodeling. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H323–H330. [Google Scholar] [CrossRef]
- Luther, D.J.; Thodeti, C.K.; Shamhart, P.E.; Adapala, R.K.; Hodnichak, C.; Weihrauch, D.; Bonaldo, P.; Chilian, W.M.; Meszaros, J.G. Absence of type VI collagen paradoxically improves cardiac function, structure, and remodeling after myocardial infarction. Circ. Res. 2012, 110, 851–856. [Google Scholar] [CrossRef]
- Grenne, B.; Eek, C.; Sjoli, B.; Dahlslett, T.; Uchto, M.; Hol, P.K.; Skulstad, H.; Smiseth, O.A.; Edvardsen, T.; Brunvand, H. Acute coronary occlusion in non-ST-elevation acute coronary syndrome: Outcome and early identification by strain echocardiography. Heart 2010, 96, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Sonaglioni, A.; Albini, A.; Fossile, E.; Pessi, M.A.; Nicolosi, G.L.; Lombardo, M.; Anza, C.; Ambrosio, G. Speckle-Tracking Echocardiography for Cardioncological Evaluation in Bevacizumab-Treated Colorectal Cancer Patients. Cardiovasc. Toxicol. 2020, 20, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J. Mol. Cell. Cardiol. 2016, 94, 189–200. [Google Scholar] [CrossRef]
- Shinde, A.V.; Frangogiannis, N.G. Fibroblasts in myocardial infarction: A role in inflammation and repair. J. Mol. Cell. Cardiol. 2014, 70, 74–82. [Google Scholar] [CrossRef]
- van Nieuwenhoven, F.A.; Turner, N.A. The role of cardiac fibroblasts in the transition from inflammation to fibrosis following myocardial infarction. Vasc. Pharmacol. 2013, 58, 182–188. [Google Scholar] [CrossRef]
- Hwang, M.W.; Matsumori, A.; Furukawa, Y.; Ono, K.; Okada, M.; Iwasaki, A.; Hara, M.; Miyamoto, T.; Touma, M.; Sasayama, S. Neutralization of interleukin-1beta in the acute phase of myocardial infarction promotes the progression of left ventricular remodeling. J. Am. Coll. Cardiol. 2001, 38, 1546–1553. [Google Scholar] [CrossRef]
- Turner, N.A.; Das, A.; Warburton, P.; O’Regan, D.J.; Ball, S.G.; Porter, K.E. Interleukin-1alpha stimulates proinflammatory cytokine expression in human cardiac myofibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1117–H1127. [Google Scholar] [CrossRef]
- Chen, B.; Frangogiannis, N.G. Chemokines in Myocardial Infarction. J. Cardiovasc. Transl. Res. 2021, 14, 35–52. [Google Scholar] [CrossRef]
- Chen, W.; Frangogiannis, N.G. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim. Biophys. Acta 2013, 1833, 945–953. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Bujak, M.; Li, N.; Gonzalez-Quesada, C.; Mendoza, L.H.; Wang, X.F.; Frangogiannis, N.G. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ. Res. 2010, 107, 418–428. [Google Scholar] [CrossRef]
- Dobaczewski, M.; de Haan, J.J.; Frangogiannis, N.G. The extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium. J. Cardiovasc. Transl. Res. 2012, 5, 837–847. [Google Scholar] [CrossRef] [PubMed]
- DeLeon-Pennell, K.Y.; Lindsey, M.L. Somewhere over the sex differences rainbow of myocardial infarction remodeling: Hormones, chromosomes, inflammasome, oh my. Expert Rev. Proteom. 2019, 16, 933–940. [Google Scholar] [CrossRef]
- Cavasin, M.A.; Sankey, S.S.; Yu, A.L.; Menon, S.; Yang, X.P. Estrogen and testosterone have opposing effects on chronic cardiac remodeling and function in mice with myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1560–H1569. [Google Scholar] [CrossRef]
- Glezeva, N.; Moran, B.; Collier, P.; Moravec, C.S.; Phelan, D.; Donnellan, E.; Russell-Hallinan, A.; O’Connor, D.P.; Gallagher, W.M.; Gallagher, J.; et al. Targeted DNA Methylation Profiling of Human Cardiac Tissue Reveals Novel Epigenetic Traits and Gene Deregulation Across Different Heart Failure Patient Subtypes. Circ. Heart Fail. 2019, 12, e005765. [Google Scholar] [CrossRef]
- Tonry, C.; Linden, K.; Collier, P.; Ledwidge, M.; McDonald, K.; Collins, B.C.; Watson, C.J. Proteomic Characterisation of Heart Failure Reveals a Unique Molecular Phenotype for Hypertrophic Cardiomyopathy. Biomedicines 2024, 12, 1712. [Google Scholar] [CrossRef]
- Zamilpa, R.; Zhang, J.; Chiao, Y.A.; de Castro Bras, L.E.; Halade, G.V.; Ma, Y.; Hacker, S.O.; Lindsey, M.L. Cardiac wound healing post-myocardial infarction: A novel method to target extracellular matrix remodeling in the left ventricle. Methods Mol. Biol. 2013, 1037, 313–324. [Google Scholar] [PubMed]
- Robinson, E.; Tate, M.; Lockhart, S.; McPeake, C.; O’Neill, K.M.; Edgar, K.S.; Calderwood, D.; Green, B.D.; McDermott, B.J.; Grieve, D.J. Metabolically-inactive glucagon-like peptide-1(9-36)amide confers selective protective actions against post-myocardial infarction remodelling. Cardiovasc. Diabetol. 2016, 15, 65. [Google Scholar] [CrossRef]
- Mouton, A.J.; Ma, Y.; Rivera Gonzalez, O.J.; Daseke, M.J., 2nd; Flynn, E.R.; Freeman, T.C.; Garrett, M.R.; DeLeon-Pennell, K.Y.; Lindsey, M.L. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res. Cardiol. 2019, 114, 6. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russell-Hallinan, A.; Tonry, C.; Kerrigan, L.; Edgar, K.; Collier, P.; McDonald, K.; Ledwidge, M.; Grieve, D.; Karuna, N.; Watson, C. Proteome Alterations in Cardiac Fibroblasts: Insights from Experimental Myocardial Infarction and Clinical Ischaemic Cardiomyopathy. Int. J. Mol. Sci. 2025, 26, 3846. https://doi.org/10.3390/ijms26083846
Russell-Hallinan A, Tonry C, Kerrigan L, Edgar K, Collier P, McDonald K, Ledwidge M, Grieve D, Karuna N, Watson C. Proteome Alterations in Cardiac Fibroblasts: Insights from Experimental Myocardial Infarction and Clinical Ischaemic Cardiomyopathy. International Journal of Molecular Sciences. 2025; 26(8):3846. https://doi.org/10.3390/ijms26083846
Chicago/Turabian StyleRussell-Hallinan, Adam, Claire Tonry, Lauren Kerrigan, Kevin Edgar, Patrick Collier, Ken McDonald, Mark Ledwidge, David Grieve, Narainrit Karuna, and Chris Watson. 2025. "Proteome Alterations in Cardiac Fibroblasts: Insights from Experimental Myocardial Infarction and Clinical Ischaemic Cardiomyopathy" International Journal of Molecular Sciences 26, no. 8: 3846. https://doi.org/10.3390/ijms26083846
APA StyleRussell-Hallinan, A., Tonry, C., Kerrigan, L., Edgar, K., Collier, P., McDonald, K., Ledwidge, M., Grieve, D., Karuna, N., & Watson, C. (2025). Proteome Alterations in Cardiac Fibroblasts: Insights from Experimental Myocardial Infarction and Clinical Ischaemic Cardiomyopathy. International Journal of Molecular Sciences, 26(8), 3846. https://doi.org/10.3390/ijms26083846